Research
Research
Abstract
A genome contains all of the genetic information of a given organism. Recent decoding of genome sequences of various animals since the turn of the century has promoted genome sciences and systems biology as the next revolution in the biological sciences. The Marine Genomics Unit (MGU) at OIST has been conducting research in four major areas, based on genome decoding: (1) Developmental and evolutionary genomics on the origin and evolution of chordates; (2) A new scientific approach toward understanding the establishment and collapse of mutualism between corals and symbiotic dinoflagellates (Symbiodinium), and crown-of-thorns starfish problem (termed environmental genomics); (3) Functional genomics of various, specialized functions that metazoans developed evolutionarily, and (4) Metazoan genome decoding projects in collaboration with zoologists throughout Japan and abroad, with the hope of sequencing genomes from all animal phyla.
(1) Developmental and evolutionary genomics on the origin and evolution of chordates
We are investigating evolutionary developmental mechanisms involved in the origin and evolution of chordates. Chordates consist of three major taxa, cephalochordates (amphioxus), urochordates (ascidians), and vertebrates (e.g., mammals). These groups share all possess notochords, dorsal, hollow neural tubes, and somites. Chordates are members of a superphyletic group of deuterostomes together with echinoderms (starfish and sea urchins) and hemichordates (acorn worms). Therefore, chordates are thought to have evolved from a common ancestor of deuterostomes.

(a) The Ciona intestinalis genome

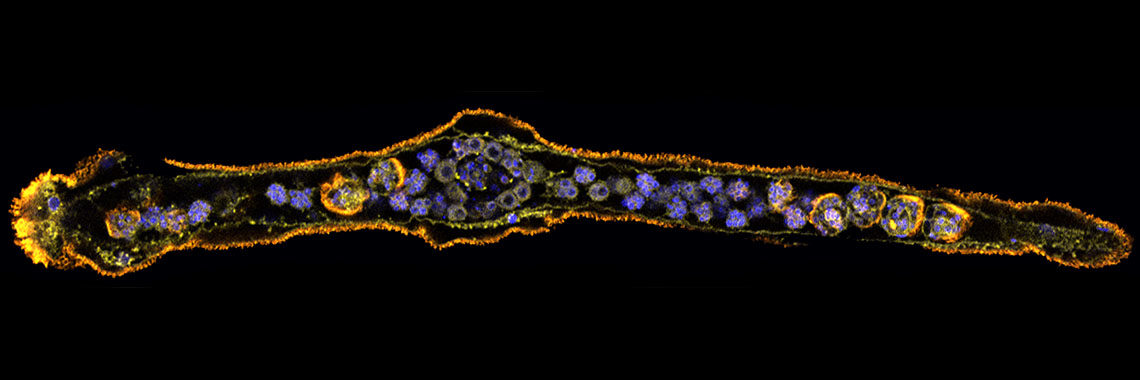
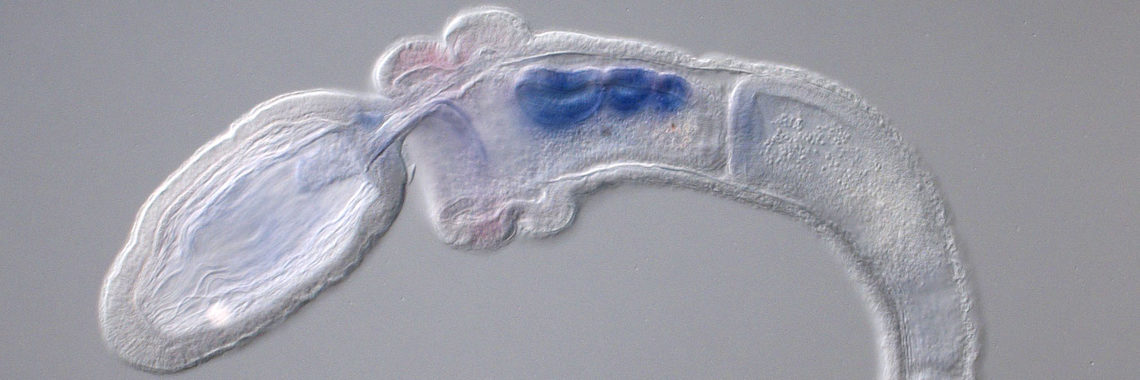
The ascidian tadpole larva represents the most simplified chordate body-plan [Satoh, Developmental Genomics of Ascidians, Wiley, (2014)]. A fertilized egg of Ciona intestinalis develops within 18 hours to a larva, which consists of approximately 2600 cells with distinct cell types, including epidermis, nervous system, muscle, notochord, mesenchyme, and endoderm. The lineages of embryonic cells are well documented. Using the decoded genome information, we have identified almost all genes for transcription factors and signaling molecules that govern ascidian body-plan formation. Taking advantage of this simple mode of embryogenesis, as well as well-characterized genes involved in developmental, Ciona is now a model experimental system for exploring gene regulatory networks for differentiation of embryonic cells [Imai et al., Science 312: 1183-1187 (2006): Shoguchi et al., Dev. Biol. 316: 498-509 (2008)].
Specifically, we found that a T-box transcription factor gene, Brachyury, plays a pivotal role in notochord development. Our group showed that Brachyury is conserved in almost all metazoans, suggesting that the acquisition of a Brachyury secondary expression domain and novel associated functions enabled Brachyury-dependent notochord formation [Satoh et al., Evol. Dev. 14: 56-75. (2012)]. In collaboration with other labs, we are now carrying out several experiments to understand the developmental mechanisms of notochord formation at the single-cell level.
The Ciona genome resolved at least two significant issues in relation to the evolution of tunicates. First, although tunicates are close relatives of vertebrates, they appear to have evolved as advanced filter-feeders, independently of the two other chordate taxa [Satoh, Zool. Sci. 26: 97-111 (2008)]. They are only animals that can synthesize cellulose by themselves. This ability derives from a gene that encodes cellulose synthase (CesA), acquired horizontally from bacteria (see section 3).
(b) The Branchiostoma floridae genome
In 2008, we decoded the genome of the amphioxus, Branchiostoma floridae [Putnam et al., Nature 453: 1064-1071 (2008)]. As a result, it became evident that, among chordates, cephalochordates diverged first, representing most basal chordates, while urochordates and vertebrates diverged later, forming a sister-group. This suggests that chordate ancestor was a free-living, vermiform-like animal. In addition, synteny is well conserved between cephalochordate and vertebrate genomes, suggesting the evolution of the chordate karyotype.
(c) The Saccoglossus kowalevskii and Ptychodera flava genomes
Around 2013, since the genome of a sea urchin (echinoderms) was sequenced in 2006, the only deuterostome group lacking a decoded genome was the hemichordates. In collaboration with a UC Berkeley research group, we finally completed this project in 2015 [Simakov et al., Nature 527: 459-464 (2015)]. During the early stage of the project, we found that two acorn worms, Saccoglossus kowalevskii and Ptychodera flava, contain a well-conserved Hox gene cluster, comparable to that of amphioxus [Freeman et al., Curr. Biol. 22: 2053–2058 (2012)]. The two acorn worm genomes showed that pterobranchs and acorn worms are members of a discrete taxon of hemichordates. We found that a cluster of four transcription factor genes, Nkx-1, Nkx2, Pax1/9, and FoxA, is expressed in the pharynx formation region of early juveniles. Since this cluster is not found in non-deuterostomes, it is highly likely that this cluster provides a cue to understand the origin and evolution of deuterostomes.

(d)
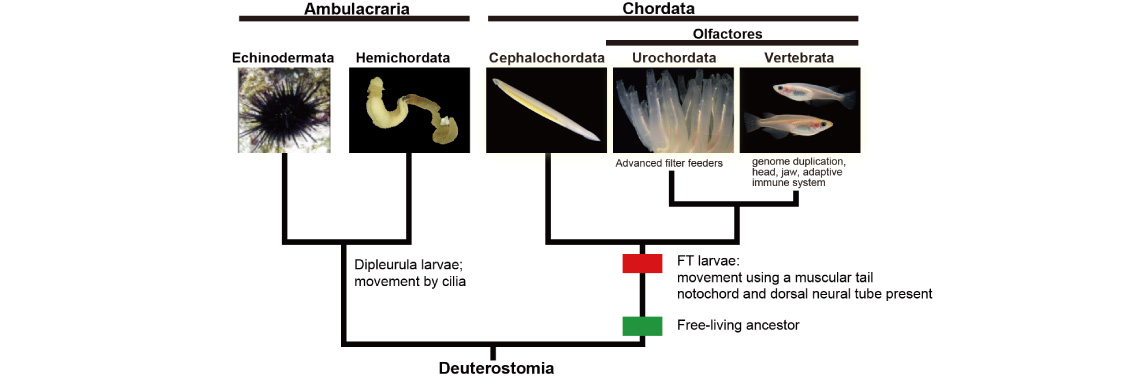
As mentioned above, we now have genomic information from all five deuterostome taxa. Comparing the genomes and data obtained from evo-devo studies of deuterostomes, we are now investigating molecular mechanisms that enabled the evolution of chordates. Traditional metazoan phylogeny classifies the Vertebrata as a subphylum of the phylum Chordata, together with two other subphyla, the Urochordata (Tunicata) and the Cephalochordata. The Chordata, together with the phyla Echinodermata and Hemichordata, comprise a major group, the Deuterostomia. Chordates invariably possess a notochord and a dorsal neural tube. Although the origin and evolution of chordates has been studied for more than a century, few authors have addressed taxonomic ranking of the three chordate groups. Accumulating evidence shows that echinoderms and hemichordates form a clade (the Ambulacraria), and that within the Chordata, cephalochordates diverged first, with tunicates and vertebrates forming a sister group.
Chordates all possess a tadpole-type larva having a notochord and a hollow nerve cord, whereas ambulacrarians have dipleurula-type larvae possessing a hydrocoel. We proposed that the development of a tadpole-type larva is fundamental to understanding mechanisms of chordate origin [Satoh, Genesis 46: 614-622 (208)]. We also suggested that the profound dipleurula versus tadpole larval differences merits a taxonomic rank higher than the phylum. Accordingly, we recommended that the Chordata should be classified as a superphylum, with the three chordate taxa as phyla [Satoh et al., Proc. Roy. Soc. B, 281: 20141729 (2015)]. A new book published by Satoh attempts to explain how chordates originated and evolved (Satoh, N. Chordate Origins and Evolution, Academic Press, 2016).
We are now pursuing comparative genomic analyses of genes involved in the origin and evolution of chordates.
Publications
- Satoh, N. Chordate Origins and Evolution. Academic Press, New Your (2016)
- Satoh, N. Developmental Genomics of Ascidians. Wiley Brackwell, New York (2014).
- Simakov, O., Kawashima, T., -------- Lowe, C.J., Satoh, N., Rokhsar, D.S., Gerhart, J., Hemichordate genomes and deuterostome origins. Nature 527(7579):459-65. (2015)
- Putnam, N.H., et al., The amphioxus genome and the evolution of the chordate karyotype. Nature 453: 1064-1071(2008).
- Dehal, P., et al., The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science 298: 2157-67 (2002)
- Imai, K.S., Levine, M., Satoh, N., and Satou, Y., Regulatory blueprint for a chordate embryo. Science 312: 1183-1187 (2006).
- Shoguchi, E., Hamaguchi, M., and Satoh, N., Genome-wide network of regulatory genes for construction of a chordate embryo. Dev. Biol. 316: 498-509 (2008).
- Satoh N., An aboral-dorsalization hypothesis for chordate origin. Genesis, 46:614-622 (2008)
- Freeman R, Ikuta T, Wu M, Koyanagi R, Kawashima T, Tagawa K, Humphreys T, Fang GC, Fujiyama A, Saiga H, Lowe C, Worley K, Jenkins J, Schmutz J, Kirschner M, Rokhsar D, Satoh N, Gerhart J. Identical Genomic Organization of Two Hemichordate Hox Clusters. Current Biology 22: 2053–2058 (2012).
- Satoh, N., Rokhsar, D., Nishikawa, T., Chordate evolution and the three-phylum system. Proc. R. Soc. B. 281:1794 (2014)
- Satoh, N., Tagawa, K., and Takahashi, H., How was the notochord born? Evolution & Development 14:56-75. (2012)
(2) Environmental genomics of coral reef biology
Although coral reefs occupy less than 0.1% of all marine habitats, they are estimated to harbor around one-third of all described marine species. Their productivity supports around one-quarter of marine fisheries. Declines in coral abundance and wholesale loss of reef habitats are one of the most pressing environmental issues of our time. The major architects of coral reefs, the scleractinian corals, are anthozoan cnidarians that form obligate endosymbioses with photosynthetic dinoflagellates of the genus Symbiodinium. The symbionts confer on the coral holobiont the ability to fix CO2 and to deposit the massive aragonite (a form of calcium carbonate) skeletons that distinguish reef-building corals from other anthozoans, such as sea anemones. The association is fragile, however, and collapses under stress from sea temperature increases, acidification, pollution, etc. Despite the ecological and economic significance of corals, molecular mechanisms underlying much of coral biology—including stress responses and disease—remain to be elucidated.

(a) The Acropora digitifera genome
Okinawa’s islands are surrounded by beautiful coral reefs, providing a superb location to study coral biology. On the assumption that genomic information of both corals and Symbiodinium are essential for understanding coral/Symbiodinium symbiosis, we selected a coral, Acropora digitifera, for genome decoding. This coral is the most abundant in Okinawa and was most severely damaged during the 1998 global coral bleaching disaster. In 2011, using next-generation sequencing technology, we sequenced the first coral genome, from A. digitifera [Shinzato et al., Nature 476: 320-323 (2011)]. This ~420-megabase genome contains approximately 23,700 gene models. Molecular phylogenetics indicate that Acropora and the sea anemone, Nematostella vectensis, diverged approximately 500 million years ago, considerably earlier than the time at which modern corals first appeared in the fossil record (240 million years ago). Despite the long evolutionary history of endosymbiosis, no evidence was found for horizontal transfer of genes from symbiont to host. However, unlike several other corals, Acropora seems to lack an enzyme essential for cysteine biosynthesis, implying the dependence of this coral on its symbiont for this amino acid. Corals inhabit environments where they are frequently exposed to high levels of solar radiation, and analysis of the Acropora genome indicates that the coral host can independently carry out de novo synthesis of mycosporine-like amino acids, which are potent ultraviolet-protective compounds. In addition, the coral innate immunity repertoire is notably more complex than that of the sea anemone, indicating that some of these genes may have roles in symbiosis or coloniality.
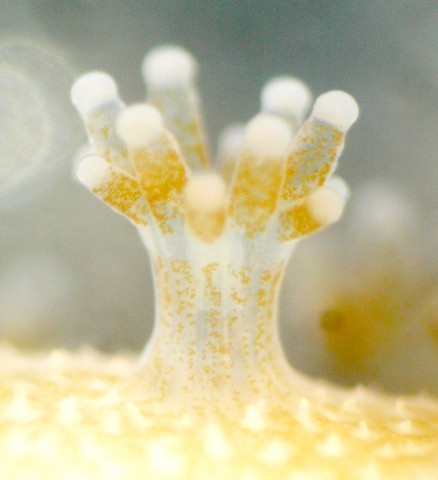
(b) Symbiodinium genome.
Then, in 2013, we succeeded in decoding a Symbiodinium genome [Shoguchi et al., Curr. Biol. 23: 1399-1408 (2013)]. The OIST Marine Genomics Unit is the lab where both coral and dinoflagellate symbiont genomes have been decoded. Dinoflagellates are ecologically and economically important unicellular eukaryotes that inhabit both marine and freshwater habitats. Most dinoflagellates are 10–100 µm in diameter and are characterized by two flagella, with a unique cell covering known as the theca. They show remarkable characteristics such as permanently condensed, liquid-crystalline chromosomes. However, dinoflagellates possess some of the largest nuclear genomes known among eukaryotes (1,500–245,000 Mbp). These huge genomes have previously thwarted whole-genome sequencing of dinoflagellates. We tackled this challenge by selecting a cultured dinoflagellate, Symbiodinium minutum, which has one of the smallest reported dinoflagellate genomes (1,500 Mbp). Sequencing reads from S. minutum were assembled into 616 Mbp gene-rich DNA regions that represented roughly half of the estimated 1,500 Mbp genome of this species. The assembly however encoded approximately 42,000 protein-coding genes, consistent with previous dinoflagellate gene number estimates using transcriptomic data. The Symbiodinium genome contains duplicated genes for regulator of chromosome condensation proteins, nearly one-third of which have eukaryotic orthologs, whereas the remainder have most likely been acquired through bacterial horizontal gene transfers. Symbiodinium genes are enriched in spliceosomal introns (mean =18.6 introns/gene). Donor and acceptor splice sites are unique, with 50 sites utilizing not only GT but also GC and GA, whereas at 30 sites, a conserved G is present after AG. Surprisingly, the Symbiodinium genome displays unidirectionally aligned genes throughout the genome, forming a cluster-like gene arrangement. At present, such gene alignment has only been seen previously in Trypanosoma genomes.
Having both coral and Symbiodinium genomes, we are now conducting experiments to analyze molecular mechanisms involved in their symbiosis.
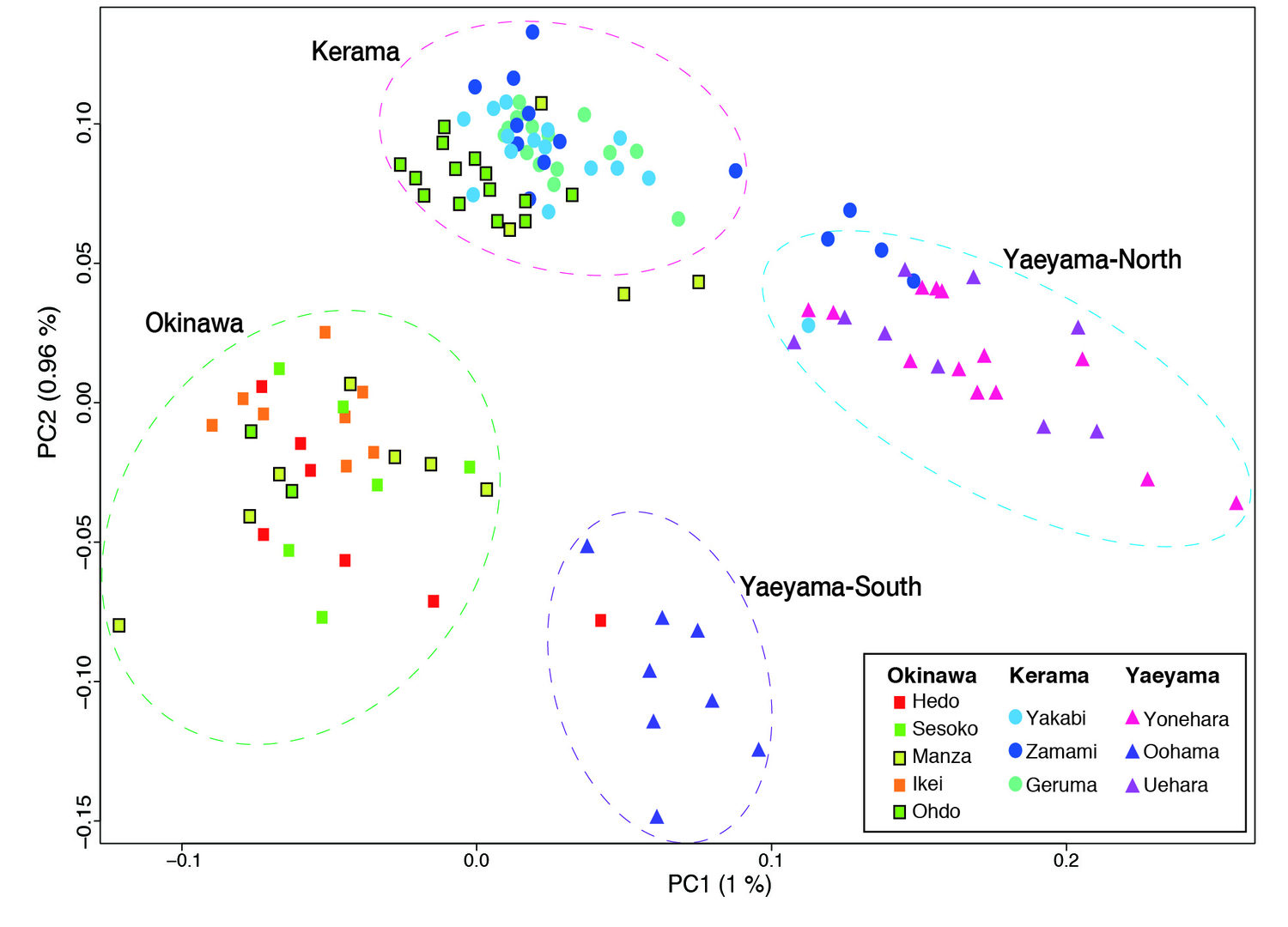 (c) Population genetics of Acropora in the Ryukyu Archipelago
(c) Population genetics of Acropora in the Ryukyu Archipelago
Taking advantage of the decoded genome of Acropora digitifera, we have analyzed the genomes of 155 samples of A. digitifera corals, collected across the Ryukyu Archipelago [Shinzato et al., Sci. Reports 5: 18211 (2015)]. We focused on subtle genetic variations that can be used to differentiate these corals into subpopulations by examining 905,561 of these variations, called single nucleotide polymorphisms or SNPs (“snips”). Genome analysis showed that these corals divide into 4 groups, corresponding to their geographical distribution: the Okinawa Islands, the Kerama Islands, and the Yaeyama-North and Yaeyama-South regions. This has significant implications regarding the recovery from the 1998 bleaching. If the Okinawa populations had been recovered due to migration of coral larvae from the Keramas, the Okinawa and Kerama clusters should show signs of genetic merging. Instead these clusters are very distinct, showing that these two populations have remained isolated. It means that long-distance larval migration from sources within the Ryukyu Archipelago is less common than previously thought. It is therefore more likely that Okinawa populations that survived the 1998 bleaching repopulated themselves, without recruited larvae from the Kerama Islands.
(d) Genetic background supporting the artificial restoration of corals
Okinawa Prefecture is now conducting a project for coral preservation and restoration. The main procedure is to promote asexual reproduction using coral branches, which are broken off healthy corals and transplanted. Population genetics data are essential to avoid mixing genetically identical colonies. Taking advantage of decoded genome information of A. digitifera and A. tenius, we have recently developed microsatellite markers that should be useful for most Acropora species. Fourteen microsatellite markers showed non-significant departure from Hardy–Weinberg equilibrium in both A. tenuis and A. digitifera. Thus these markers may provide powerful tools for population genetics studies and conservation of Acropora corals.
 (e) The Crown-of-Thorns starfish (COTS) genome
(e) The Crown-of-Thorns starfish (COTS) genome
The crown-of-thorns starfish (COTS, the Acanthaster planci species group) is a highly fecund predator of reef-building corals throughout the Indo-Pacific region1. COTS population outbreaks cause substantial loss of coral cover, diminishing the integrity and resilience of reef ecosystems. In collaboration with Australian researchers, we sequenced genomes of COTS from the Great Barrier Reef, Australia and Okinawa, Japan to identify gene products that underlie species-specific communication and could potentially be used in biocontrol strategies [Hall et al., Nature 544: 231-234 (2017)]. We focused on water-borne chemical plumes released from aggregating COTS, which make the normally sedentary starfish become highly active. Peptide sequences detected in these plumes by mass spectrometry are encoded in the COTS genome and expressed in external tissues. The exoproteome released by aggregating COTS consists largely of signalling factors and hydrolytic enzymes, and includes an expanded and rapidly evolving set of starfish-specific ependymin-related proteins. These secreted proteins may be detected by members of a large family of olfactory-receptor-like G-protein-coupled receptors that are expressed externally, sometimes in a sex-specific manner. This study provides insights into COTS-specific communication that may guide the generation of peptide mimetics for use on reefs with COTS outbreaks.
In the process of COTS genome decoding project, we noticed that the genome sequencing assembly became of a high quality, which makes us to investigate the gene constituent in echinoderm genomes. As a result, we found the presence in the COTS genome of an organized Hox cluster, exception of lacking Hox6, the result of which is contrast to a dis-organized Hox cluster in the sea urchin genome [Baughman et al., genesis 52: 952-958 (2014)]. In addition, the Nkx2 cluster (see 1c) is found to be intact in the COTS genome, although extant echinoderms lost pharyngeal gills.
Furthermore, an interesting finding of COTS genome was an extreme similarity of genome sequences between GBR and OKI individuals, which were collected from location ~5,000 km apart. We are now examining COTS population genetics in relation to their vast occurrence.
Publications
- Hall, M.R., Kocot, K.M., Baughman, K.W., Fernandez-Valverde, S.L., Gauthier, M.E.A., Hatleberg, W.L., Krishnan, A., McDougall, C., Motti, C.A., Shoguchi, E., Wang, T., Xiang, X., Zhao, M., Bose, U., Shinzato, C., Hisata, K., Fujie, M., Kanda, M., Cummins, S.F., Satoh, N., Degnan, S.M., Degnan. B.M. The crown-of-thorns starfish genome as a guide for biocontrol of this coral reef pest. Nature, 544: 231-234 (2017).
- Shinzato, C., Shoguchi, E., Kawashima, T., Hamada, M., Hisata, K., Tanaka, M., Fujie, M., Fujiwara, M., Koyanagi, R., Ikuta, T., Fujiyama, A., Miller, D. J., and Satoh, N. Using the Acropora digitifera genome to understand coral responses to environmental change. Nature, 476: 320-323 (2011).
- Shoguchi, E., Shinzato, C., Kawashima, T., Gyoja, F., Mungpakdee, S., Koyanagi, R., Takeuchi, T., Hisata, K., Tanaka, M., Fujiwara, M., Hamada, M., Azadeh, S., Fujie, M., Usami, T., Goto, H., Yamasaki, S., Arakaki, N., Suzuki, Y., Sugano, S., Toyoda, A., Kuroki, Y., Fujiyama, A., Medina, M., Coffroth, M. A., Bhattacharya, D., and Satoh, N. Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Current Biology 23: 1399-1408 (2013).
- Shinzato, C., Mungpakdee, S., Arakaki, N., Satoh, N. Genome-wide SNP analysis explains coral diversity and recovery in the Ryukyu Archipelago. Scientific Reports 5, Article number: 18211 (2015) doi:10.1038/srep18211
- Shinzato, C., Yasuoka, Y., Mungpakdee, D., Arakaki, A., Fujie, M., Nakajima, Y., Satoh, N. Development of novel, cross-species microsatellite markers for Acropora corals using next-generation sequencing technology. Front. Mar. Sci., doi : 10.3389/fmars. 2014.00011 (2014).
- Baughman KW, McDougall C, Cummins SF, Hall M, Degnan BM, Satoh N, Shoguchi E. Genomic organization of Hox and ParaHox clusters in the echinoderm, Acanthaster planci. Genesis 52: 952-8. (2014).
(3) Functional genomics
(a) Cellulose biosynthetic pathway
Cellulose represents the biggest biomass on the earth. Development and/or improvement of methods for its effective use are essential to maintain human life. Cellulose biosynthesis takes place at the so-called terminal complex, embedded in the cell membrane, which makes it hard to elucidate its molecular mechanisms. Cellulose synthase (CesA) and cellulase are the major proteins involved in this process. At the time of the Ciona genome decoding project, we found that C. intestinalis contains a single copy of CesA (Ci-CesA), which is expressed in epidermal cells of developing tailbud embryos [Nakashima et al., Dev. Genes Evol. 214: 81-88 (2004)]. Dr. Yasunori Sasakura and his colleagues have established Ciona transgenic lines using the Minos transposon. A line named swimming juveniles (sj) is a mutant that lacks the ability to synthesize cellulose, in which Minos is inserted into 5’ upstream sequences of Ci-CesA, resulting in a failure of gene function [Sasakura et al., PNAS USA102, 15134-15139 (2005)]. On the other hand, a larvacean tunicate, Oikopleura dioica, contains two copies of Ci-CesA, each of which encodes a different crystal structure of cellulose. Taking advantage of these tunicate systems, we are now conducting studies to explore molecular mechanisms of cellulose biosynthesis in tunicates.

(b) The pearl oyster genome.
Bivalve molluscs have flourished in marine environments, and many species constitute important aquatic resources. Especially pearl oysters produce pearls, one of the most economically valuable fishery products. In 2012, we decoded the ~1 Gb genome of the pearl oyster, Pinctada fucata [Takeuchi et al., DNA Res. 19: 117-130 (2012)]. In addition, we published an improved genome assembly of P. fucata (version 2.0) [Takeuchi et al., DNA Res. 19: 117-130 (2012)] by updating the assembly of approximately 193 coverage of the genome. Gene model version 2.0 was generated with the aid of manual gene annotations supplied by the P. fucata research community. Comparison of mollusc and other bilaterian genomes shows that gene arrangements of Hox, ParaHox, and Wnt clusters in the P. fucata genome are similar to those of other molluscs. A number of tandem duplications of genes that encode shell matrix proteins are well characterized in the P. fucata genome. The genomic data are now being used to characterize genes that are involved in biomineralization.
 (c) Seaweed genomics
(c) Seaweed genomics
The brown alga, Cladosiphon okamuranus (Okinawa mozuku), is economically one of the most important edible seaweeds, and is cultivated for market primarily in Okinawa, Japan. C. okamuranus constitutes a significant source of fucoidan, which has various physiological and biological activities. To facilitate studies of seaweed biology, we decoded approximately 140-Mb genome of C. okamuranus S-strain [Nishitsuji et al., DNA Research 23: 561-570 (2016)], which is estimated to contain 13,640 protein-coding genes, approximately 94% of which have been confirmed with corresponding mRNAs. Comparisons with the other brown algal genome identified a set of C. okamuranus genes that encode enzymes involved in biosynthetic pathways for sulfated fucans and alginate biosynthesis. We would like to use the Cladosiphon genome for future studies of mozuku biology.
Publications
- Nakashima, K., Yamada, L., Satou, Y., Azuma, J.-i. and Satoh, N.: The evolutionary origin of animal cellulose synthase. Dev. Genes Evol. 214, 81-88 (2004).
- Sasakura, Y., Nakashima, K., Awazu, S., Matsuoka, T., Nakayama, A., Azuma, J. and Satoh, N.: Transposon-mediated insertional mutagenesis revealed the functions of animal cellulose synthase in the ascidian Ciona intestinalis. Proc. Natl. Acad. Sci. USA 102, 15134-15139 (2005).
- Takeuchi, T., Kawashima, T., Koyanagi, R., Gyoja, F., Tanaka, M., Ikuta, T., Shoguchi, E., Fujiwara, M., Shinzato, C., Hisata, K., Fujie, M., Usami, T., Nagai, k., Maeyama, K., Okamoto, K., Aoki, H., Ishikawa, T., Masaoka, T., Fujiwara, A., Endo, K., Endo, H., Nagasawa, H., Kinoshita, S., Asakawa, S., Watabe, S., and Satoh, N., Draft Genome of the Pearl Oyster Pinctada fucata: A Platform for Understanding Bivalve Biology. DNA Research 19: 117-130. (2012)
- Takeuchi, T., Koyanagi, R., Gyoja, F., Kanda, M., Hisata, K., Fujie, M., Goto, H., Yamasaki, S., Nagai, K., Morino, Y., Miyamoto, H., Endo, K., Endo, H., Nagasawa, H., Kinoshita., S., Asakawa, S., Watabe, S., Satoh, N., Kawashima, T., Bivalve-specific gene expansion in the pearl oyster genome: Implications of adaptation to a sessile lifestylei. Zoological Letters, 2:3 (2016)
- Nishitsuji, K., Arimoto, A., Iwai, K., Sudo, Y., Hisata, K., Fujie, M., Arakaki, N., Kushiro, T., Konishi, T., Shinzato, C., Satoh, N., Shoguchi, E. A draft genome of the brown alga, Cladosiphon okamuranus, S-strain: a platform for future studies of 'mozuku' biology. DNA Research 23: 561-570 (2016).
(4) Genome decoding project of metazoans
OIST is a new graduate school, launched in 2011. The power of its genome sequencing facility (DNA Sequencing Section: SQC) however has supported genome projects of various organisms. Taking advantage of this resource and considering my Unit’s 20-year contribution the zoological sciences, we decided to undertake the challenge of decoding genomes of all metazoan phyla not yet decoded. Since genomes of all deuterostome phyla, and most diploblast animal phyla, unsequenced phyla include most Spilaria and/or Lophotrochozoa. This project involves collaborations with many zoologists in Japan and foreign countries. In 2015, we succeeded in decoding a Brachiopod genome (below), and we are now close to finishing those of the Phoronida and the Nemertini. On-going targets are the Acoela, Mesozoa, Gastrotricha, Entoprocta, Bryozoa, Chaetognatha, and others.
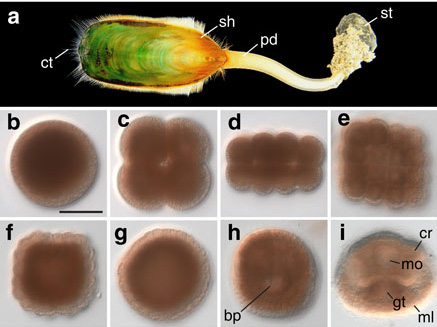
(a) The Lingula anatina genome:
The evolutionary origins of lingulid brachiopods and their calcium phosphate shells have attracted researchers for many years. They are sometimes called “living fossils.” We decoded the 425-Mb genome of Lingula anatina to gain insights into brachiopod evolution [Luo et al., Nature Comm. 6: 8301 (2015)]. Comprehensive phylogenomic analyses place Lingula close to molluscs, but distant from annelids. The Lingula gene number increased to approximately 34,000 by extensive expansion of gene families. Although Lingula and vertebrates have superficially similar hard tissue components, our genomic, transcriptomic, and proteomic analyses show that Lingula lacks genes involved in bone formation, indicating an independent origin of their phosphate biominerals. Several genes involved in Lingula shell formation are shared by molluscs. However, Lingula has independently undergone domain combinations to produce shell matrix collagens with EGF domains, and it carries lineage-specific shell matrix proteins. This Lingula genome provides resources for further studies of lophotrochozoan evolution.
Publications
- Luo, Y.J., Takeuchi, T., Koyanagi, R., Yamada, L., Kanda, M., Khalturina, M., Fujie, M., Yamasaki, S., Endo, K., Satoh, N.,The Lingula genome provides insights into brachiopod evolution and the origin of phosphate biomineralization. Nature Communications, 18;6:8301. doi: 10.1038/ncomms9301 (2015).
News Feed
Followings are the research productivities of our unit for this fiscal year, which were all posted on OIST News Feed.
Please see the News Feed to check details for each article.