

FY2020 Annual Report
Chemistry and Chemical Bioengineering Unit
Professor Dr. Fujie Tanaka, Professor
Abstract
The ability to design and synthesize organic molecules constitutes the foundation that underlies basic research as well as applied science. The ability is also essential for the development of pharmaceuticals and biofunctional molecules. This unit develops efficient, concise, and safe chemical transformation methods and strategies for constructing small molecules bearing functional groups and/or chiral centers that are relevant to the creation of biofunctional molecules. We design and create small organic molecule catalyst systems for designed chemical transformations, and we develop reaction strategies that use small organic molecules as enzyme-like catalysts. With the use of organic molecules as catalysts, we minimize the need for protection and deprotection steps that are usually required for the synthesis of functionalized molecules. When a reaction method does not affect functional groups that are not at the reaction site, the reaction method can be used for the synthesis of a series of molecules bearing various functional groups. This means that a series of molecules of interest may be synthesized using the same method in a short route, providing advantages for the synthesis of biofunctional candidate molecules. We also investigate the chemical bases of the reactions to understand the mechanisms of the catalysis and molecular interactions provided by organic molecules. By taking advantage of the use of features of our developing molecules, we also develop strategies and methods for conjugation of proteins and peptides with other molecules. The research undertaken by this unit advances the chemistry of catalysis and of molecular synthesis. The studies by this unit accelerate the creation of molecules used in biomedical research and contribute to the development of new therapeutics, therapeutic strategies, and diagnostic methods.
1. Staff
- Dr. Ravindra D. Aher
- Dr. Venkati Bethi
- Dr. Santosh Chavan
- Dr. Yuvraj Garg
- Dr. Yarkali Krishna
- Dr. Prodip M. Roy
- Dr. Muhammad Sohail
- Kola Shilpa
- Santanu Mondal, Graduate Student
- James Osborne, Graduate Student
- Maira Pasha, Special Research Student
- Tatiana Gridneva, Graduate Student (Rotation)
-
Shiho Chinen, Research Unit Administrator
2. Activities and Findings
2.1. Development of new chemical transformation methods and synthesis of functionalized molecules
We have been developing small organic molecule catalysts (organocatalysts) and organocatalytic molecular transformation methods useful for the synthesis of functionalized molecules under mild conditions in short routes. We also investigate the chemical bases of the catalyses and the chemical transformations to understand the mechanisms of the catalysis and molecular interactions provided by organic molecules to further the creation of useful molecules.
Traditional synthetic methods often require high or very low temperatures and/or absolute conditions. In addition, functional groups on substrate molecules must be protected prior to reactions. That is, depending on functional groups present in target molecules to be synthesized, synthetic routes, including protection and deprotection steps, have to be designed for each molecule. To concisely synthesize functionalized molecules, chemical transformation methods that are not affected by functional groups presenting in starting materials are needed. It is a great advantage when the same reaction method can be used for the synthesis of a series of molecules bearing various functional groups without the need of product-specific protection and deprotection steps. In addition, it is desired that such reactions can be performed under safe, mild, and environmentally benign conditions. We address these points in our research as we design and develop catalysts and chemical transformation methods. By using organic molecules as catalysts, we concisely synthesize novel functionalized molecules including those that are often difficult to synthesize by traditional synthetic strategies. Our investigations into the chemical basis of the developed catalysts and chemical transformation methods further the understanding of the chemistry of organic molecules and their reactions.
2.1.1. Enantioselective direct anti-selective Mannich reactions catalyzed by 3-pyrrolidinecarboxylic acid in the presence of potassium carbonate
Asymmetric Mannich reactions of ketones with imines are important transformations to synthesize enantiomerically enriched amino ketones, amino alcohols, and their related derivatives. Various types of catalysts including metal-based catalysts and organocatalysts have been used for providing the Mannich products with high diastereo- and enantioselectivities. However, each catalyst system has a certain scope. To expand or alter the scope, different catalysts are usually required. The design and the synthesis of new catalysts are important for the development of reaction methods, but we sought an alternative way to expand the scope and to improve stereoselectivities of the reactions catalyzed by certain catalysts. We have demonstrated that the use of K2CO3 as additive improves the enantioselectivities of the reactions catalyzed by 3-pyrrolidinecarboxylic acid with retaining the anti-selectivities (Scheme 1) (Yuvraj and Tanaka, Org. Lett. 2020, 22, 4542).

Scheme 1
Homochiral 3-pyrrolidinecarboxylic acid (or β-proline) has been used for catalyzing direct Mannich-type reactions of ketones or aldehydes with N-p-methoxyphenyl (PMP)-protected imine of glyoxylates to afford anti-isomers of Mannich products with high diastero- and enantioselectivities. However, the 3-pyrrolidinecarboxylic acid-catalyzed reactions of cyclohexanone with PMP-protected imines of arylaldehydes previously provided the anti-isomers of the Mannich products with low or moderate enantioselectivities. Previously, few examples of the reactions that afford the anti-isomers of the Mannich products from cyclohexanone and PMP-protected imines of arylaldehydes with high diastereo- and enantioselectivities have been reported.
In the previously reported 3-pyrrolidinecarboxylic acid-catalyzed reactions of ketones or aldehydes with the imine of ethyl glyoxylate, the hydrogen bonds between the carboxylic acid of the catalyst and the imine or the hydrogen transfer from the carboxylic acid to the imine can locate the glyoxylate imines at the position suitable for the C-C bond formation and control the stereochemistries of the products, leading to the formation of the anti-Mannich products (Figure 1a). In contrast, in the same catalyst-catalyzed reactions of ketones with the imines of arylaldehydes, the hydrogen bonds or the hydrogen transfer between the carboxylic acid and the imines may not locate the imines at the positions suitable for the C-C bond formation or may locate the imines at the position a little far from that suited for the C-C bond formation (Figure 1b). Accordingly, the C-C bond formation may occur without the involvement of the hydrogen bonds or the hydrogen transfer, resulting the formation of the products with moderate to low enantioselectivities. The differences between the reactions of the ethyl glyoxylate imine and of the arylaldehydes imines may originate from the electron-withdrawing feature and/or the structure of the ester group of the glyoxylate imine. The electron-withdrawing group may affect the length of the hydrogen bonds in the transition state. We hypothesized that addition of alkali metal salts, alkaline earth metal salts, and other additives (such as non-transition metals and ammonium salts) would tune the positioning of the imines in the transition state for the C-C bond formation, leading to the product formation with high enantioselectivities while retaining the anti-selectivity (Figure 1c). Then, we found that addition of K2CO3 improved the enantioselectivities of the Mannich reactions of six-membered ring ketones with the N-PMP-protected imines of arylaldehydes catalyzed by 3-pyrrolidinecarboxylic acid. The O-K and the N-K bonds are longer than the O-H and the N-H bonds in the O-H-N hydrogen bond; the longer bonds would allow the positioning of the reactants in the transition state for the C-C bond formation to form the product in a stereocontrolled way. We proposed that K2CO3 is involved in positioning of the imine in the transition state for the C-C bond formation to lead to the anti-Mannich product with high enantioselectivity.
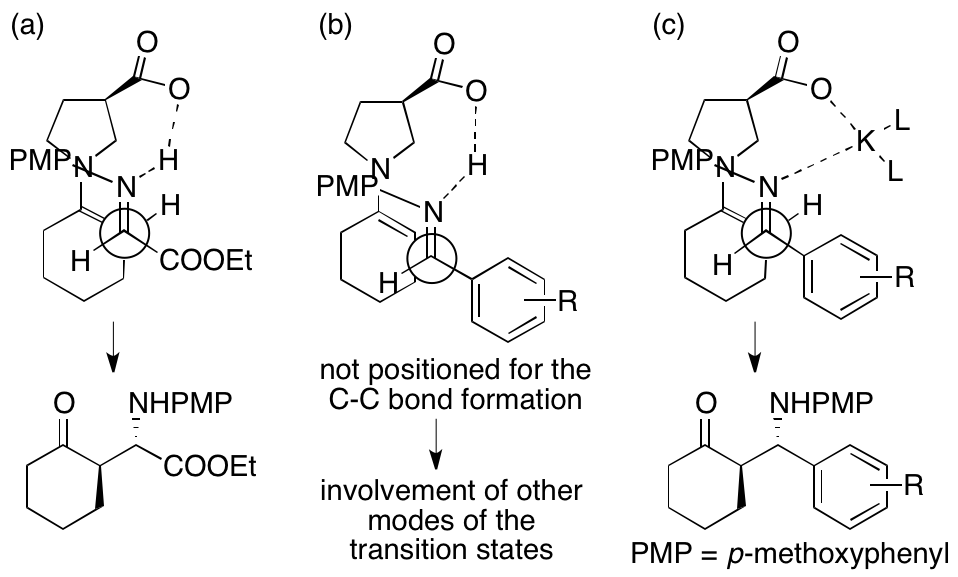
Figure 1
2.1.2. Construction of spirooxindole polycyclic derivatives
Spirooxindole derivatives are found in bioactive natural products. Functionalized molecules with spirooxindole cores including those with polycyclic systems should be useful in drug discovery efforts. We have recently reported formal (4+1) cycloaddition and enantioselective Michael-Henry cascade reactions that provide spirooxindole polycycles via spiro[4,5]decane derivatives (Scheme 2) (Huang, Sohail, Taniguchi, Monde, and Tanaka, Angew. Chem. Int. Ed. 2017, 56, 5853). The reactions provided spirooxindole all-carbon polycyclic derivatives with seven stereogenic centers, including two all-carbon chiral quaternary centers and one tetrasubstituted chiral carbon center.
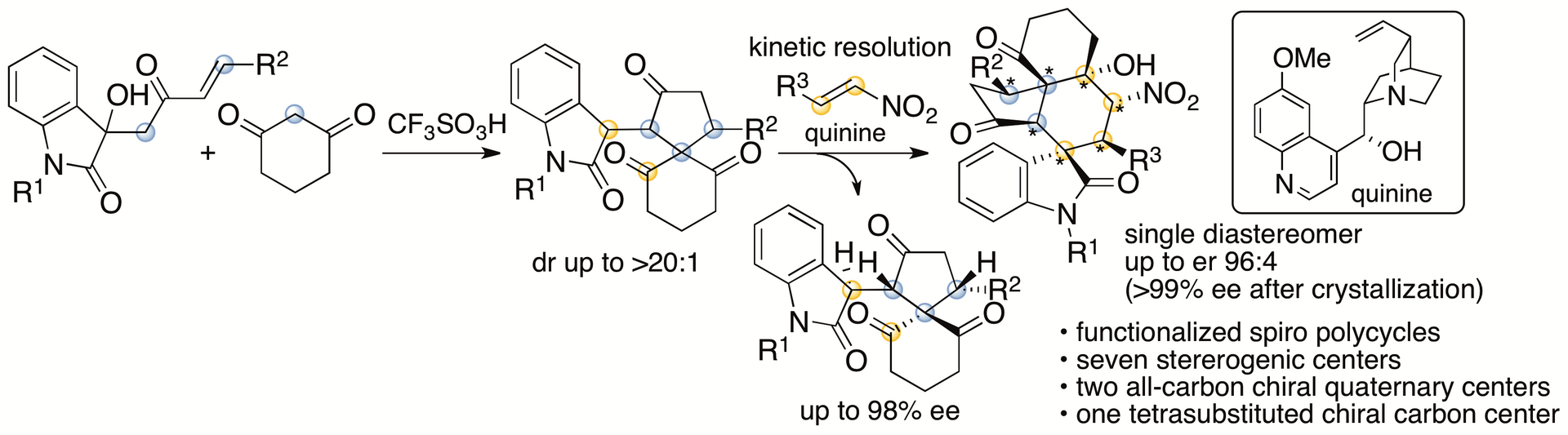
Scheme 2
We have also developed annulation via aldol-oxa-cycliztion cascade reactions to afford spirooxindole pyran polycycles from the same spiro[4,5]decane derivatives (Scheme 3) (Sohail and Tanaka, Communications Chemistry 2020, 2, article number 73). With the use of arylglyoxal derivatives as reactants, the formation of C-C and C-O bonds was achieved. Whereas the all-carbon polycycles mentioned above had a trans-cis relationship for the generated 5-6-6 ring system, the pyran polycycles generated by this method had a cis-cis relationship for the 5-6-6 ring system. During the reaction, the spiro[4,5]decane derivative was isomerized to the diastereomer, and this was the key to afford the product. From the spiro[4,5]decane derivatives obtained by kinetic resolutions, highly enantiomerically enriched single diastereomers of the spirooxindole pyran polycycles were obtained.
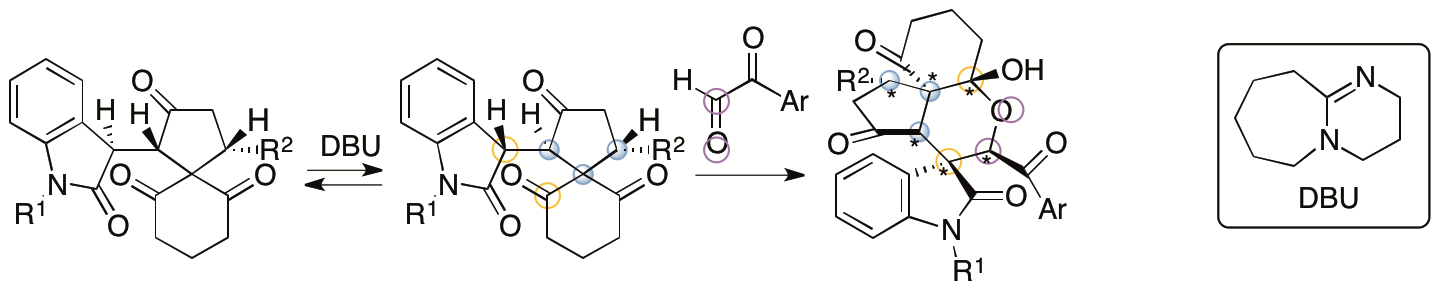
Scheme 3
Usually, these complex, polycyclic molecules are synthesized by multi step routes. Our strategies allowed the access to the complex functionalized molecules including those as highly enantiomerically enriched forms in a short route.
We have been investigating the mechanisms of the reactions reading to the formation of the spiro[4,5]decane derivatives. We are also investigating to provide other spiro polycyclic products and to expand the capabilities for the synthesis of functionalized molecules with spiro ring and polycyclic ring systems.
2.1.3. Construction of spirooxindole tetrahydropyrans: oxa-Diels-Alder reactions and oxa-Michael reactions
We previously reported catalytic asymmetric hetero-Diels-Alder reactions of enones with isatins (2,3-dioxyindoles or 2,3-dioxyindolins) that provide functionalized spirooxindole tetrahydropyranones with high diastereo- and enantioselectivities (Scheme 4) (Cui and Tanaka, Chem. Eur. J. 2013, 19, 6213). Novel amine-based catalyst systems composed of three-types of molecules (amine, acid, and thiourea) were developed to catalyze the reactions. The design and synthesis of single-molecule catalysts that provide all the required interactions for the catalysis and stereocontrol for designed reactions especially for new reactions are often difficult. We have demonstrated that the use of multicomponent catalyst systems provides a way to bypass the limitations in the access to efficient single component catalysts. We also elucidated the mechanism of the reactions and key factors for the high diastero- and enantioselectivities achieved by the three-component catalyst system (Cui, Chouthaiwale, Yin, and Tanaka, Asian J. Org. Chem. 2016, 5, 153).
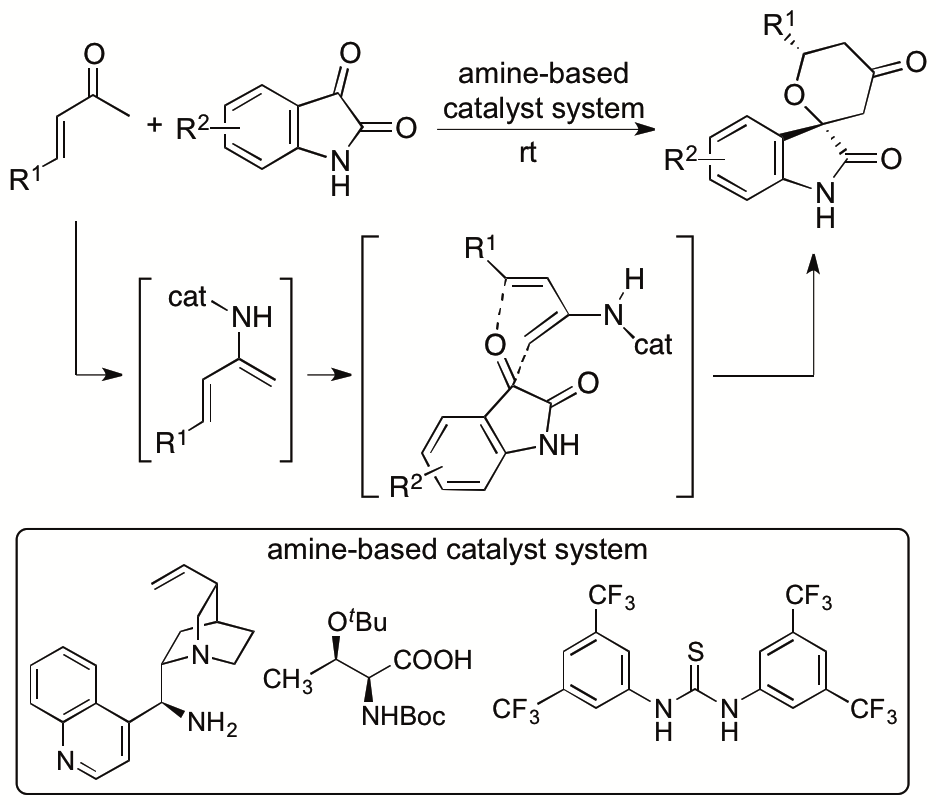
Scheme 4
We have also been expanding the oxa-hetero-Diels-Alder reactions of enones beyond the use of the oxindole dienophiles to the use of various ketones and aldehydes as dienophiles. For example, we have developed diastereo- and enantioselective hetero-Diels-Alder reactions of enones with aryl trifluoromethyl ketones (Scheme 5) (Pasha and Tanaka, Heterocycles in press, doi: 10.3987/COM-20-S(K)22). With our newly developed catalyst system, tetrahydropyranones were obtained with high diastereo- and enantioselectivities, and the major diastereomers of the products had the relative stereochemistry different from that of the reactions that we have previously reported (Zhang and Tanaka, RSC Adv. 2016, 6, 61454).
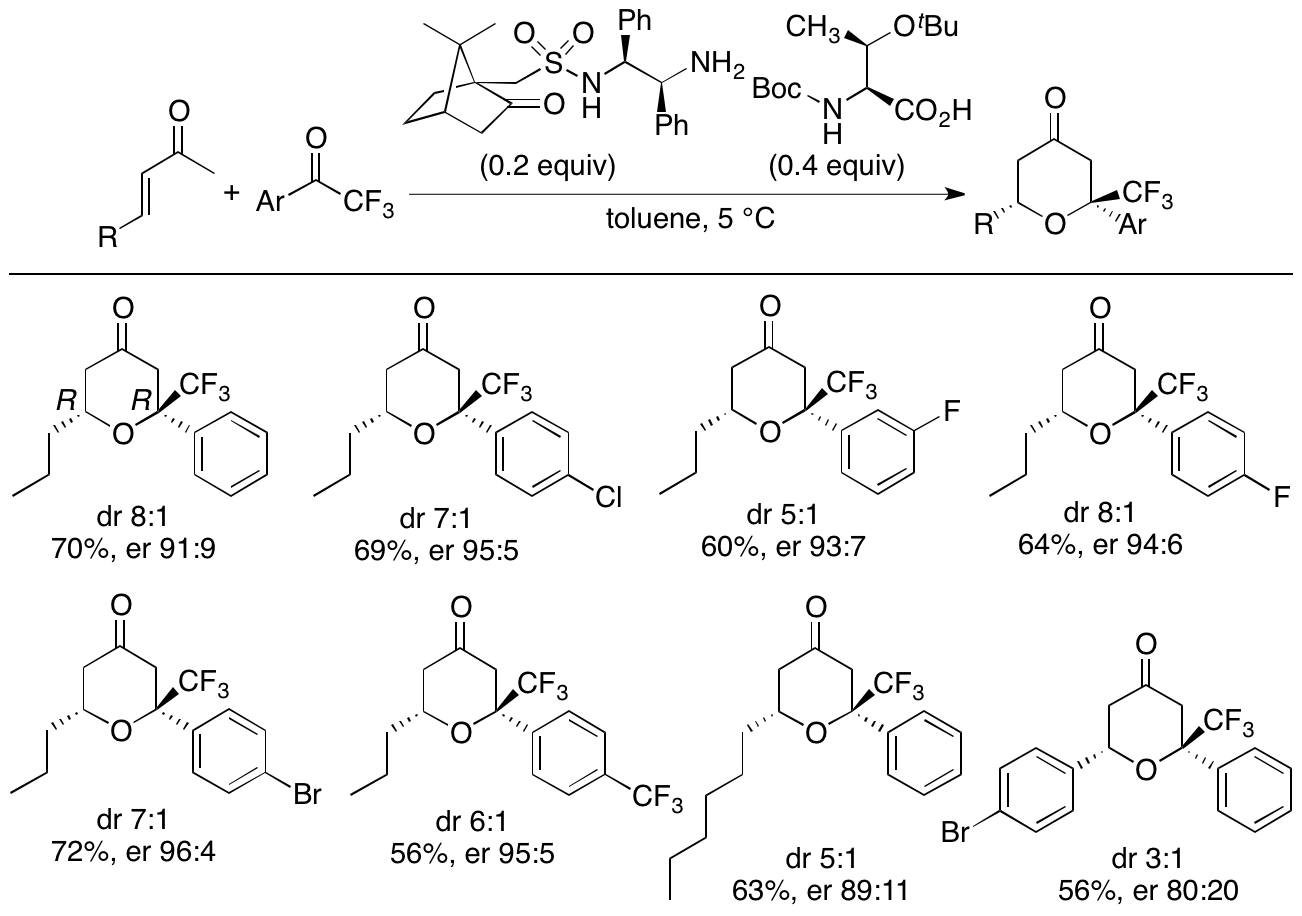
Scheme 5
We have been further studying and developing diastereo- and enantioselective hetero-Diels-Alder reactions and other strategies in order to allow the concise access to functionalized various tetrahydropyran derivatives with highly diastereo- and enantioselective manners.
2.1.4. Construction of highly functionalized decalin derivatives: aldol-aldol annulation reactions
We have recently developed catalytic enantioselective formal (4+2) cycloaddition reactions of dihydropyran derivatives with cyclohexane-1,3-diones that afford functionalized decalins through aldol-aldol annulation (Scheme 6) (Chouthaiwale, Aher, and Tanaka, Angew. Chem. Int. Ed. 2018, 57, 13298). The decalin ring system is found in diterpenes, diterpenoids, steroids, and other bioactive molecules, so the development of methods for the synthesis of functionalized decalin derivatives is of interest in drug discovery and related research. Our strategy enabled the construction of polyfunctionalized decalins bearing five to six chiral carbon centers with high diastereo- and enantioselectivities from achiral molecules in a single transformation that formed two C-C bonds.
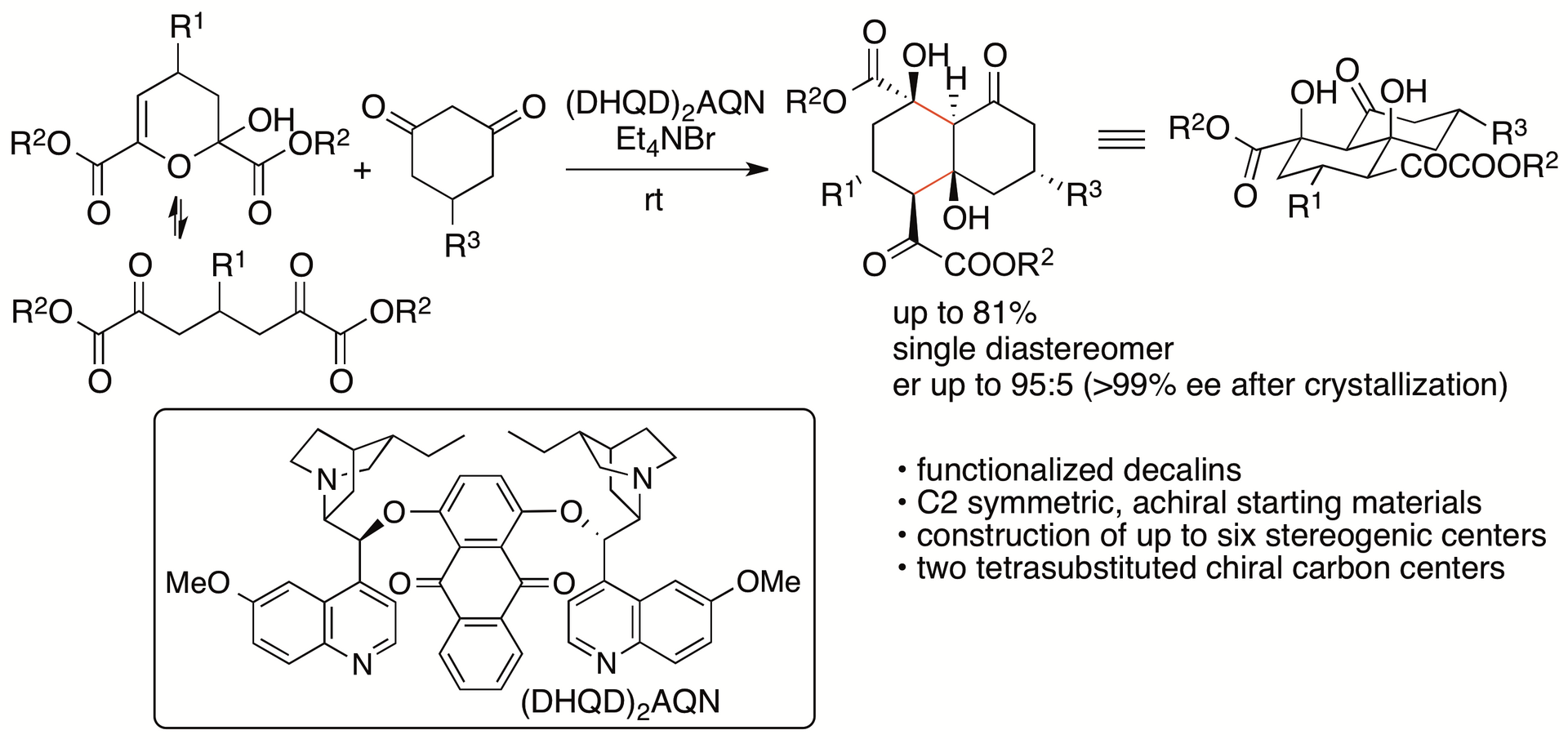
Scheme 6
Key factors of the success of the reactions include the use of the dihydropyran derivatives as the starting materials, which are obtained from pyruvates and aldehydes (Chouthaiwale and Tanaka, Chem. Commun. 2014, 50, 14881; Chouthaiwale, Lapointe, and Tanaka, Heterocycles 2017, 95, 587). The starting material dihydropyran derivatives retain the α-ketoester group of pyruvates and are C2 symmetric molecules, allowing direct enantioselective transformations to provide the products in high yields (in contrast to that kinetic resolutions result in yielding the products in no higher than 50%).
We are currently investigating the detail mechanisms of the reactions. We are also investigating essential parts of the catalyst structure for the catalysis and the stereocontrol.
2.1.5. Reactions of pyruvates to synthesize various functionalized molecules
Pyruvates can act as nucleophiles and electrophiles and thus are expected to be useful synthons. However, the dual reactivities of pyruvates are difficult to control. As described in section 2.1.4., we have demonstrated the synthesis of functionalized decalin derivatives through the reactions of pyruvates (Chouthaiwale, Aher, and Tanaka, Angew. Chem. Int. Ed. 2018, 57, 13298). We are further developing transformation methods that use the dihydropyrans obtained from pyruvates and aldehydes to concisely provide enantiomerically enriched, polyfunctionalized molecules by taming the reactivities of pyruvate derivatives.
We are also developing catalyst systems that enable the reactions of pyruvates to provide functionalized molecules and the transformation methods with the catalysts. For example, we are developing enantioselective Mannich reactions of pyruvates, in which the pyruvates act as nucleophiles (i.e., reactions of enolates and/or enamines of pyruvates). The Mannich products of pyruvates would be useful for the synthesis of α,γ-diamino acid derivatives, γ-amino α-hydroxy acid, and other functionalized molecules.
2.1.6. Constructions of spiro isoindolinones: Intramolecular formal [4+2] cycloaddition reactions
Isoindolinone derivatives are found in bioactive natural products. Methods for the synthesis of these molecules are of interests in drug discovery efforts. We have previously reported that the N-acyliminium ions generated from hydroxylactams in situ can be transformed to N-acylenamides in situ, which act as nucleophiles to react with electrophiles (Scheme 7) (Krishna, Y., Shilpa, K., and Tanaka, F. Org. Lett. 2019, 21, 8444). From hydroxylactam enals as the starting materials, depending on the substituents, the C-C bond formation for the ring formation occurred through either intramolecular Mannich reactions of the N-acyliminium ion generated in situ or intramolecular Michael reactions of the enamide also generated in situ.
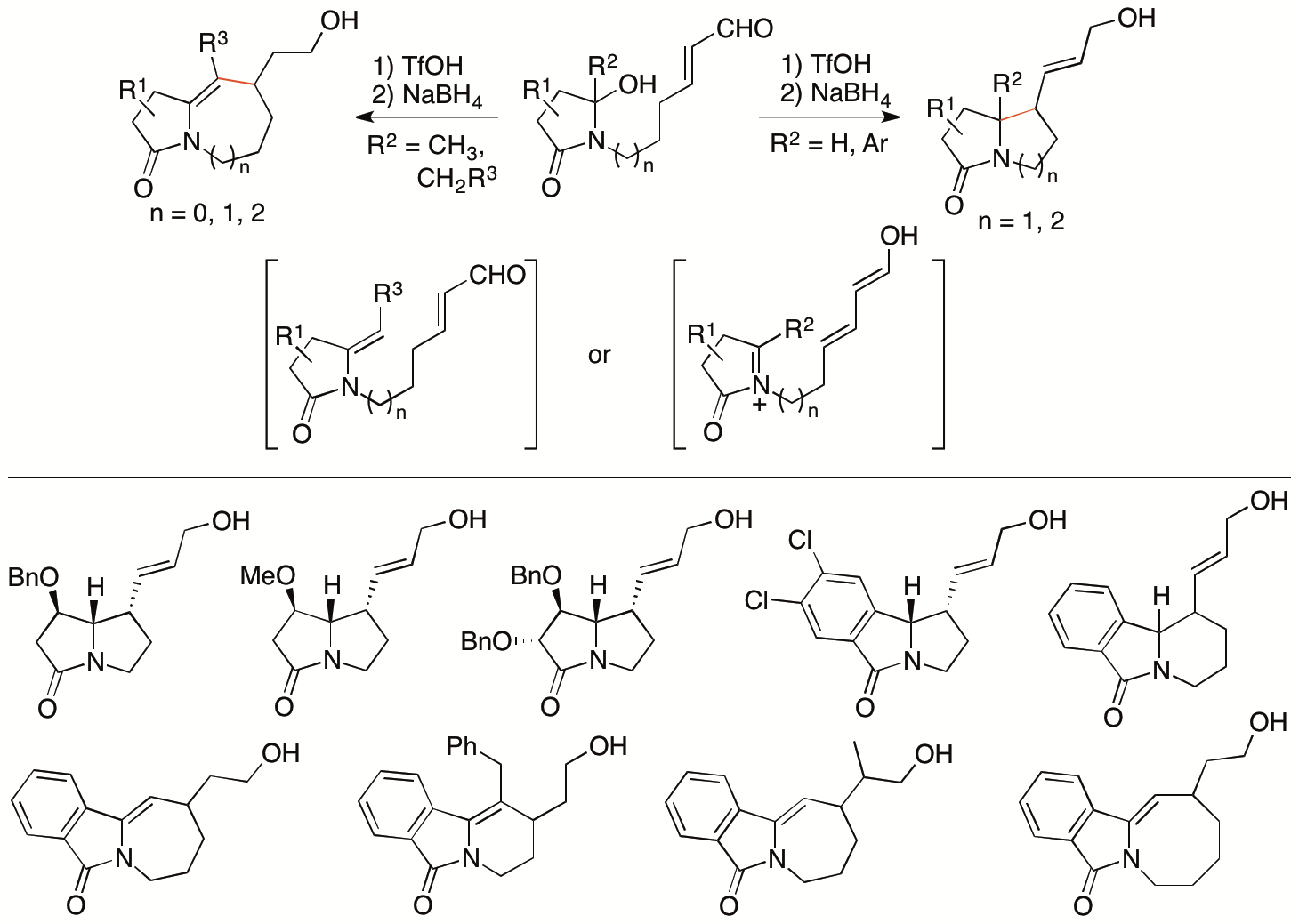
Scheme 7
By expanding the strategy of the reactions of the N-acylenamides generated in situ, we have developed intramolecular formal [4+2] cycloaddition reactions of the N-acylenamides generated in situ from isoindolinone-derived hydroxylactam derivatives to synthesize spiro isoindolinone derivatives and related isoindolinone derivatives (Scheme 8) (Krishna, Y. and Tanaka, F. Org. Lett. 2021, 23, 1874). In these reactions, two new rings are formed with the construction of the spiro ring system. The pathways would be Michael-Mannich reactions or inverse-electron demand Diels-Alder reactions. The C-O bond formed is cleaved and stable products are obtained.

Scheme 8
2.1.7. γ-Position selective reactions of β-ketoesters
In most aldol and Mannich reactions of β-ketoesters in which the β-ketoesters are used as nucleophiles, the bond formation occurs at the α-position of the β-ketoesters. Previously, we developed γ-selective aldol reactions of β-ketoesters with aryl trifluoromethyl ketones catalyzed by 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (Scheme 9) (Zhang and Tanaka, Adv. Synth. Catal. 2015, 357, 3458).
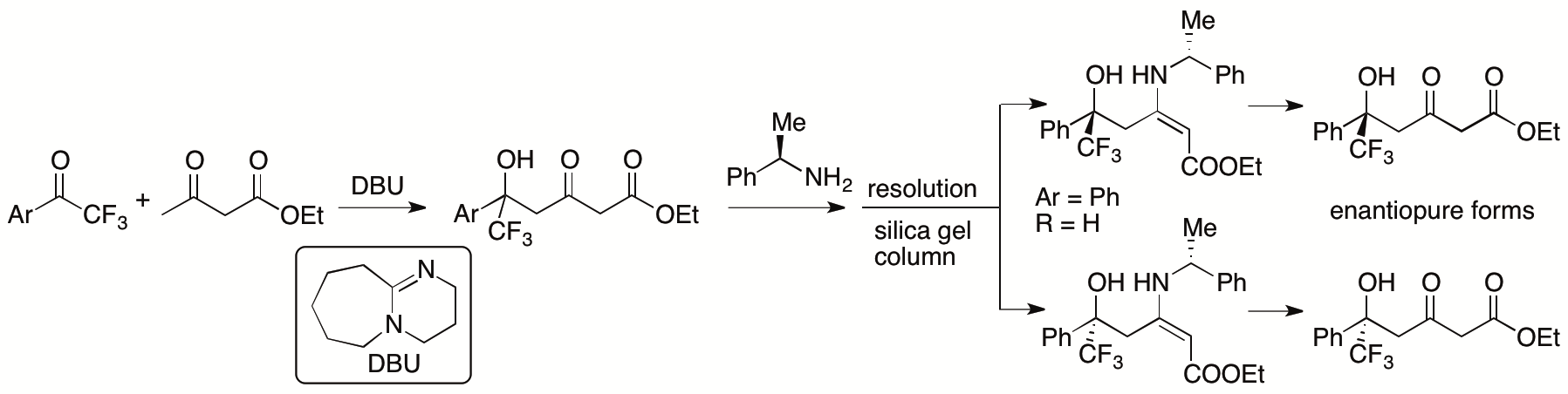
Scheme 9
Whereas resolutions of the γ-selective aldol products provided the enantiopure forms, enantioselective γ-selective aldol reactions of β-ketoesters are more desired than the resolution for the access to the enantiomerically enriched aldol products. In collaborating Prof. Dongxin Zhang, former PhD student of the Tanaka Unit, we have developed γ-selective aldol reactions of β-ketoesters with isatins (Scheme 10) (Zhang, Chen, Cai, Yin, Zhong, Man, Zhang, Bethi, and Tanaka, Org. Lett. 2020, 22, 6).
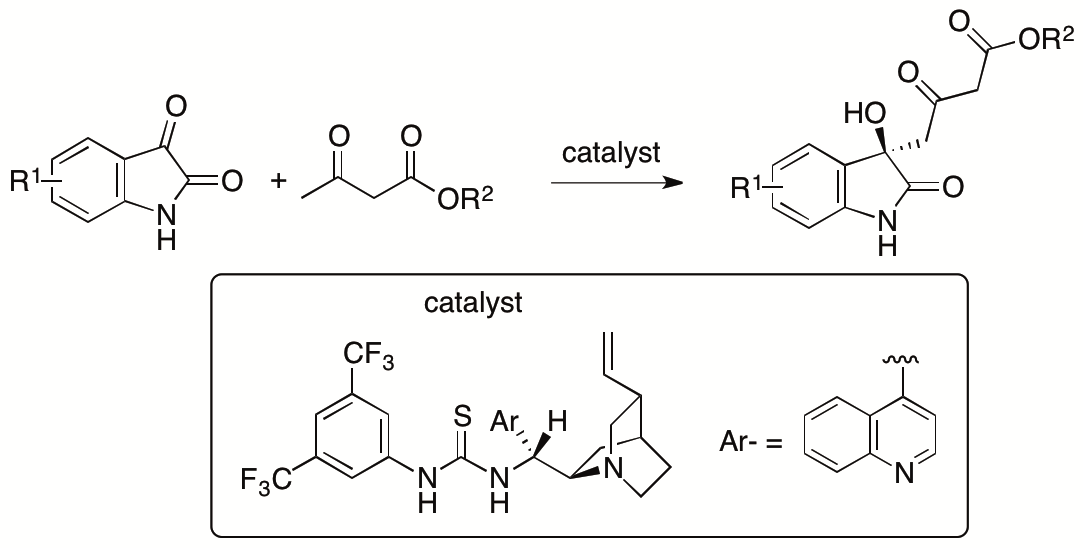
Scheme 10
We are also working to expand the γ-selective aldol reactions of β-ketoesters to those with other acceptor substrates (i.e., other than isatines). We are also developing enantioselective γ-selective Mannich and other reactions of β-ketoesters.
2.1.8. Control of chemical reactions using 1,3-cyclohexanedione derivatives as buffering molecules in non-aqueous solutions
Control of chemical reactions is necessary to obtain desired chemical transformation products and to prevent decompositions and isomerizations of molecules of interest. To control chemical reactions in aqueous solutions or to maintain conditions suitable for enzyme-catalyzed reactions and for storage of biological samples such as enzymes and antibodies, the use of buffers is a common practice. We have introduced the "buffering" concept into the events that occur in non-aqueous solutions and have demonstrated that 1,3-cyclohexanedione derivatives have buffering functions in non-aqueous solutions (Scheme 11) (Sohail and Tanaka, Chem. Eur. J. 2020, 26, 222). 1,3-Cyclohexanedione derivatives inhibited both acid-catalyzed and base-catalyzed isomerizations and decompositions in organic solvents. To suppress decompositions and isomerizations of the molecules by using the buffering molecules that we have disclosed, there is no need to consider whether the decomposition and/or isomerization is caused by a base or an acid or which type of base or acid is causing the decomposition, isomerization, and/or racemization. Additionally, simply adding the buffering molecule to reaction mixtures can completely alter the pathways of the reactions. No such simple operations to control chemical reactions were commonly demonstrated previously. The use of buffering molecules that work in organic solvents provides a strategy to control chemical reactions and expands the range of compounds that can be synthesized.
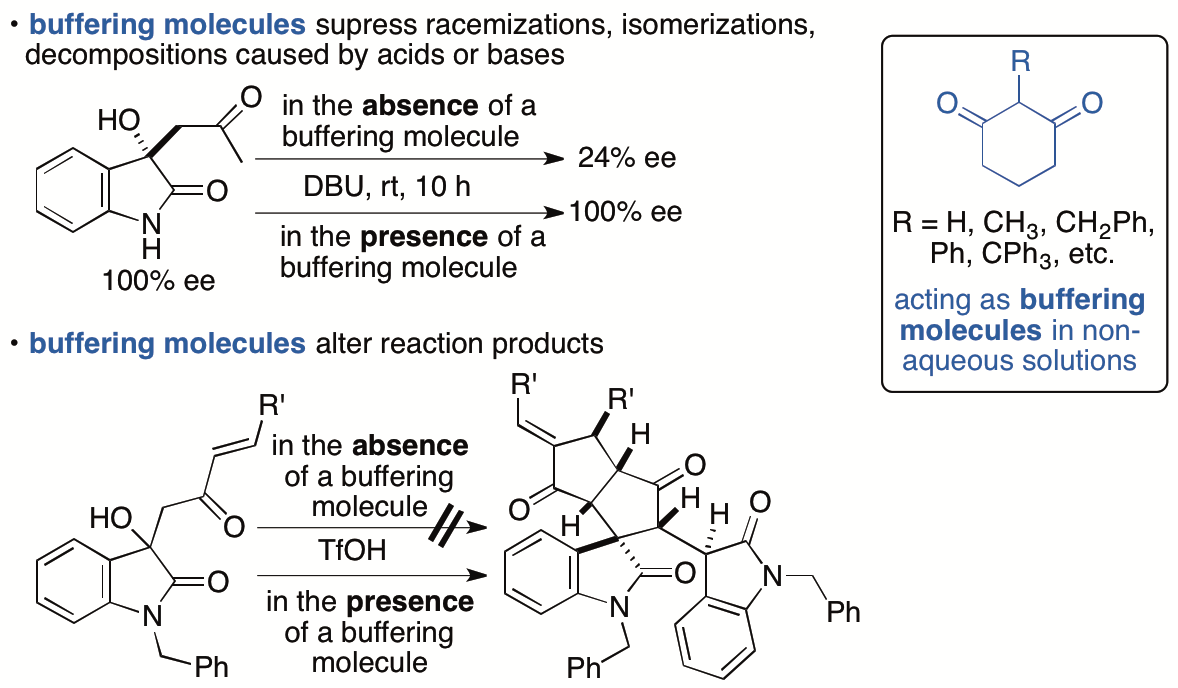
Scheme 11
We are expanding the concept for tuning the reactivities in organic solvents. We are also developing reaction systems and catalyst systems that use the buffering functions of 1,3-cyclohexanedione derivatives.
2.2. Development of bioconjugation systems
Protein labeling methods are required for the synthesis of antibody-drug conjugates and other protein conjugates; these molecules are important as therapeutics and as detection devices for molecules of interest. Conjugation reactions are also needed to create multifunctional molecules. We are developing efficient protein labeling systems and molecules with desired reactivities that can be used for protein labeling reactions at targeted sites.
2.3. Search of biofunctional molecules
As described above, we have synthesized various functionalized molecules. In collaboration with researchers whose expertise is in biology and screening for biofunctional molecules, we have been searching new biofunctional molecules and drug leads.
3. Publications
3.1 Journals
1. Garg, Y.; Tanaka, F. Enantioselective direct anti-selective Mannich-type reactions catalyzed by 3-pyrrolidinecarboxylic acid in the presence of potassium carbonate: Addition of potassium carbonate improves enantioselectivities. Organic Letters 2020, 22, 4542-4546, doi: 10.1021/acs.orglett.0c01561.
URL: https://pubs.acs.org/articlesonrequest/AOR-IT8GIC72CDMACWRSQPBY
or URL: https://pubs.acs.org/doi/full/10.1021/acs.orglett.0c01561
2. Pasha, M.; Tanaka, F. Catalytic enantioselective oxa-hetero-Diels-Alder reactions of enones with aryl trifluoromethyl ketones: Synthesis of tetrahydropyranones. Heterocycles in press, doi: 10.3987/COM-20-S(K)22.
URL: https://www.heterocycles.jp/newlibrary/libraries/doi
(then, may need to search by doi)
3. Krishna, Y.; Tanaka, F. Intramolecular Formal [4+2] cycloadditions: Synthesis of spiro isoindolinone derivatives and related molecules. Organic Letters 2021, 23,1874-1879, doi: 10.1021/acs.orglett1c00283.
URL: https://pubs.acs.org/articlesonrequest/AOR-6ZHTFSVMIUSDE37BBTT2
or URL: https://pubs.acs.org/doi/full/10.1021/acs.orglett.1c00283
3.2. Oral and Poster Presentations
1. Sohail, M.; Tanaka, F. Annulation via dynamic aldol-oxa-cyclization to construct spirooxindole pyran polycycles, in The 141st Annual Meeting of the Pharmaceutical Society of Japan, Hiroshima, Japan (2021), 2021.03.26-2021.03.29. (poster No. 28P01-005)
2. Bethi, V.; Tanaka, F. Enantioselective organocatalytic γ-position-selective Mannich reactions of β-ketoesters, in The 141st Annual Meeting of the Pharmaceutical Society of Japan, Hiroshima, Japan (2021), 2021.03.26-2021.03.29. (poster No. 28P01-001)
3. Mondal, S.; Tanaka, F. Organocatalytic enantioselective Mannich reactions of pyruvates as nucleophiles, in The 141st Annual Meeting of the Pharmaceutical Society of Japan, Hiroshima, Japan (2021), 2021.03.26-2021.03.29. (poster No. 28P01-006S)
4. Intellectual Property Rights
1. Tanaka, F.; Sohail, M. 2-Substituted 1,3-cyclohexanone derivatives as buffering molecules in non-aqueous solutions, patent application PCT/JP2020/038871, filed on 2020.10.15.