

FY2011 Annual Report
Genomics and Regulatory Systems Unit
Assistant (Adjunct) Professor Nicholas Luscombe
Abstract
Cellular life must recognise and respond appropriately to diverse internal and external stimuli. By ensuring the correct expression of specific genes at the appropriate times, the transcriptional regulatory system plays a central role in controlling many biological processes: these range from cell cycle progression and maintenance of intracellular metabolic and physiological balance, to cellular differentiation and developmental time-courses. Numerous diseases result from a breakdown in the regulatory system and a third of human developmental disorders have been attributed to dysfunctional transcription factors (TFs). Furthermore, alterations in the activity and regulatory specificity of TFs are now established as major sources for species diversity and evolutionary adaptation. Indeed, increased sophistication of the regulatory system appears to have been a principal requirement for the emergence of metazoan life.
Much of our basic knowledge of transcriptional regulation has derived from molecular biological and genetic investigation. In the past decade, the availability of genome sequences and development of new laboratory techniques have generated (and continue to generate) information describing the function and organisation of regulatory systems on an unprecedented scale. Genomic studies now allow us to examine regulatory systems from a whole-organism perspective; on the other hand however, many observations made with these data are unexpected and appear to complicate our view of gene expression control.
The continued flood of biological data means that many interesting questions require the application of computational methods to answer them. The combination of computational biology and genomics enables us to uncover general principles that apply to many different biological systems; any unique features of individual systems can then be understood within this broader context.
My group pursues research dedicated to understanding on a genomic scale: (i) how transcription is regulated and (ii) how this regulatory system controls biologically interesting phenomena. Much of our work until now has been purely computational, either analysing publicly available data or in collaboration with experimental laboratories performing functional genomic investigations.
1. Staff
- Nicholas M. Luscombe, Assistant Professor (Adjunct)
- Dr Garth Ilsley, Research Scientist
- Dr Takeshi Noda, Researcher
- Ms Sachiko Blank, Administrative Assistant
2. Collaborations
- Theme: Statistical models of transcriptional regulation in developing fly embryos
- Type of collaboration: Joint research
- Researchers: Assistant Professor, Angela DePace, Harvard Medical School
- Theme: Single cell analysis of transcriptional regulation in developing chordate embryos
- Type of collaboration: Joint research
- Researchers: Professor, Nori Satoh, OIST Graduate University
3. Activities and Findings
3.1 Statistical models of transcriptional regulation in developing fly embryos
Garth Ilsley, Nicholas Luscombe in collaboration with Angela DePace
Transcriptional control ensures that genes are expressed in the right amounts at the correct times and locations. A challenge is to understand quantitatively how regulatory systems precisely convert input signals to the appropriate outputs: what is the regulatory input function that determines a target gene’s expression pattern? Making use of the Virtual Embryo dataset from the Berkeley Drosophila Transcription Network Project, we successfully and accurately modeled the expression of even skipped (eve) stripes 2 and 3+7 across the entire embryo at cellular resolution. We showed that a straightforward statistical relationship explains how the measured concentrations of transcription factors (TF) define the complex spatial pattern of eve expression, without the need for pairwise interactions or cross-regulatory dynamic processes. By simulating outputs from thousands of TF combinations, we recovered known regulators and also suggested roles for new candidates. Finally, our models predicted the intricate effects of regulatory perturbations including mutations in regulating TFs and misexpression experiments with remarkable accuracy. In contrast to many previous methods, we imposed minimal assumptions on models; instead we identified those that best fit the data and then infer the underlying mechanistic features of the regulatory input function, such as the lack of TF-specific thresholds or the need for pairwise interactions, and the positional value of homotypic interactions. Overall, the approach provides a generally applicable and quantitative approach for elucidating the regulation of diverse biological systems and developing experimentally testable hypotheses.
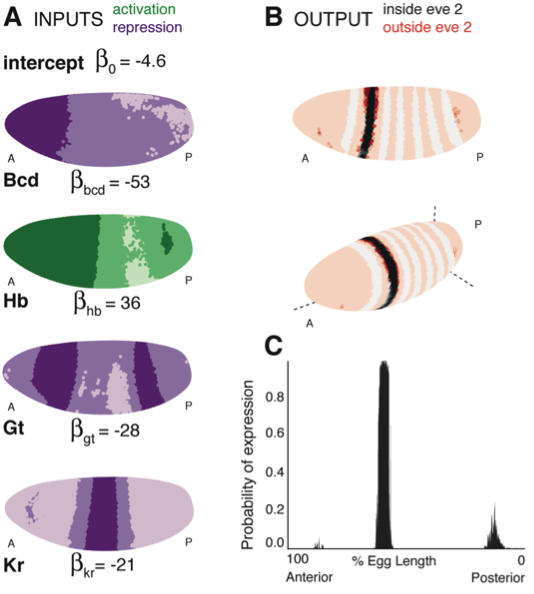
Figure 1. (A) Lateral perspectives of the Drosophila embryo depicting the contribution of four regulators (Bcd, Hb, Gt and Kr) to the model output. Embryos are drawn with the anterior (A) to the left and posterior (P) to the right, along with regulator names and corresponding coefficients in the model. Each nucleus is shaded to indicate the level of contribution by regulators, with darker colors signifying stronger effects (in this case, due to higher regulator concentrations): green represents a positive, activating effect and purple a negative, repressive one. Inputs are continuous, but drawn using a discrete color scale for simplicity. (B) Lateral and 3D perspectives of the embryo show the model prediction of eve stripe 2 expression. Each nucleus is colored from light to dark for low to high probability of eve being ON: within stripes the color scale is from white to black and outside the stripes it is on a red scale, with peach for values below 0.15. (C) A ribbon plot showing the probability of eve expression (y-axis) for nuclei within 10μm of the lateral midline along the anteroposterior axis (x-axis). The plot demonstrates that the stripe borders are sharply defined. It also allows easy comparisons with other models that are generally performed in one dimension.
3.2 Single cell analysis of transcriptional regulation in developing chordate embryos
Garth Ilsley, Takeshi Noda, Nicholas Luscombe in collaboration with Nori Satoh
Until now, molecular biology techniques have been limited in the ability to measure gene expression levels in single cells, meaning that only average measurements for entire embryos were available. In collaboration with Nori Satoh’s unit, we are developing approaches to measure gene expression and thus model transcriptional regulatory processes in individual cells, as they differentiate into specific cell types. Since many of the regulatory circuits in ciona, an invertebrate marine organism, are conserved in vertebrates, we will be able to apply many of the findings to human development.
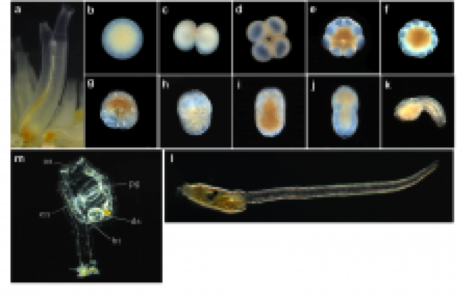
Figure 2. Images showing different stages of development in the ciona embryo.
4. Publications
4.1 Journals
- Conrad T, Cavalli FM, Holz H, Hallacli E, Kind J, Ilik I, Vaquerizas JM, Luscombe NM, Akhtar A. The MOF chromobarrel domain controls genome-wide H4K16 acetylation and spreading of the MSL complex. Dev Cell. 2012 Mar 13;22(3):610-24.
- Zaugg JB, Luscombe NM. A genomic model of condition-specific nucleosome behavior explains transcriptional activity in yeast. Genome Res. 2012 Jan;22(1):84-94.
- König J, Zarnack K, Luscombe NM, Ule J. Protein-RNA interactions: new genomic technologies and perspectives. Nat Rev Genet. 2012 Jan 18;13(2):77-83.
- Vaquerizas JM, Teichmann SA, Luscombe NM. How do you find transcription factors? Computational approaches to compile and annotate repertoires of regulators for any genome. Methods Mol Biol. 2012;786:3-19.
- Koepke J, Kaffarnik F, Haag C, Zarnack K, Luscombe NM, König J, Ule J, Kellner R, Begerow D, Feldbrügge M. The RNA-binding protein Rrm4 is essential for efficient secretion of endochitinase Cts1. Mol Cell Proteomics. 2011 Dec;10(12):M111.011213.
- Seshasayee AS, Luscombe NM. Comparative genomics suggests differential deployment of linear and branched signaling across bacteria. Mol Biosyst. 2011 Nov;7(11):3042-9.
- Cavalli FM, Bourgon R, Vaquerizas JM, Luscombe NM. SpeCond: a method to detect condition-specific gene expression. Genome Biol. 2011 Oct 18;12(10):R101.
4.2 Books and other one-time publications
Nothing to report
4.3 Oral and Poster Presentations
Nothing to report
5. Intellectual Property Rights and Other Specific Achievements
Nothing to report
6. Meetings and Events
Nothing to report
7. Other
Nothing to report.