

FY2020 Annual Report
Algorithms for Eco and Evo Genomics
Professor (Adjunct) Eugene Myers

Guarav Agavekar (Ph.D. student, Economo Unit)
Tracey Audisio (Ph.D. student, Bouguignon Unit)
Takeshi Takeuchi (Staff Scientist, Satoh Unit)
Yoshihiko Suzuki (Postdoc, Myers Unit)
(foreground inset):
Gene Myers
Abstract
Despite COVID preventing Myers onsite presence at OIST, a great deal was accomplished via electronic means and thanks to the great work of the SQC. Multi-genome, high-fidelity sequencing projects are now well underway with the Economo and Bouguignon units to study the independent, convergent evolution of certain striking traits in insects. And together with the Satoh unit a nearly perfect, phased reconstruction of the pearl oyster genome has been produced along with several corals crucial to the investigation of reef health. In addition to collaborative sequencing projects, Myers and his postdoc Suzuki have developed a novel and highly accurate software framework for evaluating k-mers in the context of genome assembly.
1. Staff
- Dr. Eugene Myers, Adjunct Professor
- Dr. Yoshihiko Suzuki, Postdoctoral Scholar
- Yukiko Murabayashi, Research Unit Administrator
2. Collaborations
2.1 Convergent Evolution of Trapjaws in Strumigenys
- Type of collaboration: Joint research
- Researchers:
- Professor Evan Economo, OIST
2.2 Convergent Evolution of Symbiosis in Beetles living among Termite Colonies
- Type of collaboration: Joint research
- Researchers:
- Assistant Professor Thomas Bourguignon, OIST
2.3 A Hapltype-base Population Study of Crown of Thorns Starfish and Their Ecosystem
- Type of collaboration: Joint research
- Researchers:
- Professor Noriyuki Satoh, OIST
2.4 Towards a perfect rconstruction of the iconic Medaka Fish
- Type of collaboration: Joint research
- Researchers:
- Professor Shinishi Morishito, U. Tokyo
3. Activities and Findings
3.1 Convergent Evolution of Trapjaws in Strumigenys
The genus Strumigenys is a hyperdiverse clade of 950+ species of predatory ants distributed globally throughout the tropics. In order to capture prey with rapid escape mechanisms, the Economo group has shown previously that ultrafast trap-jaw mandibles developed 7-10 times independently in different regions of the world. We seek to understand the molecular basis of this phenotype and the mechanism by which it evolved using a comparative genomics approach.
To this end we are sequencing 10-15 selected species within the genus, as well as outgroups, that can then be mined for both proteomic and regulatory hypotheses. We have produced near perfect, phased reconstructions of about half the collection at this point in time using a combination of low-input HiFi long-read sequencing and Omni-C paired reads for scaffolding. A particular obstacle has been the low yield of DNA obtained from individuals, where by we sequence bar-coded DNA from 2-3 larvae.
Next year we aim to complete the sequence and reconstruction phase and will move into annotation, comparison, and also digital transcript expression if the data reveals that the most likely evolutionary mechanism was changes in gene regulation.
3.2 Convergent Evolution of Symbiosis in Beetles living among Termite Colonies
Within the clade of rove beetles are many species that have have developed obligate symbiotic relationships with ants or termites. There is further a range of phenotypic variations evolved to facilitate these relationships such as the development of a protective shell (luminoid) or host-like body plans (phasogastric). These adaptions have also occurred convergently several times within the clade. Like the Economo collaboration, we have been planning to take a comparative genomic approach using onwards of 30-40 species. The project is just getting underway, but in 2021 we hope to be nearly finished with the sequencing and reconstruction phase of the project.
3.3 Reference Sequencing of Iconic Japanese Animals
Before beginning the multi-genome projects described immediately above, we first tested and developed our new sequencing protocols at the SQC on a number of important Japanese animals. Together with the Satoh group, we used a combination of Omni-C and HiFi long reads to produce a chromosome level, near complete, phased reconstruction of the pearl oyster. There in we were able to see surprisingly large, transposon-induced variation between haplotypes affecting immune system fucntion.
Looking to produce even more continuous, potentially telomere-to-telomere reconstrutions, we have produced ultra-long nanopore reads for several species of Medaka fish. With the Morishita group at U. Tokyo we are evaluating the value of this data to close all the gaps in high quality reconstructions such as the pearl oyster above.
3.4 Algorithmic Methods for Genome Assembly
This year I personally developed the fastest algorithm and software for counting k-mers in high-fidelity, shotgun DNA data sets called FastK. I further built superior versions of several, much-used k-mer analysis tools compatible with FastK. Together this suite of apps is being incorporated into the Sanger Center's sequencing operations pipeline, and hopefully it will be adopted at many other major sequencing centers.
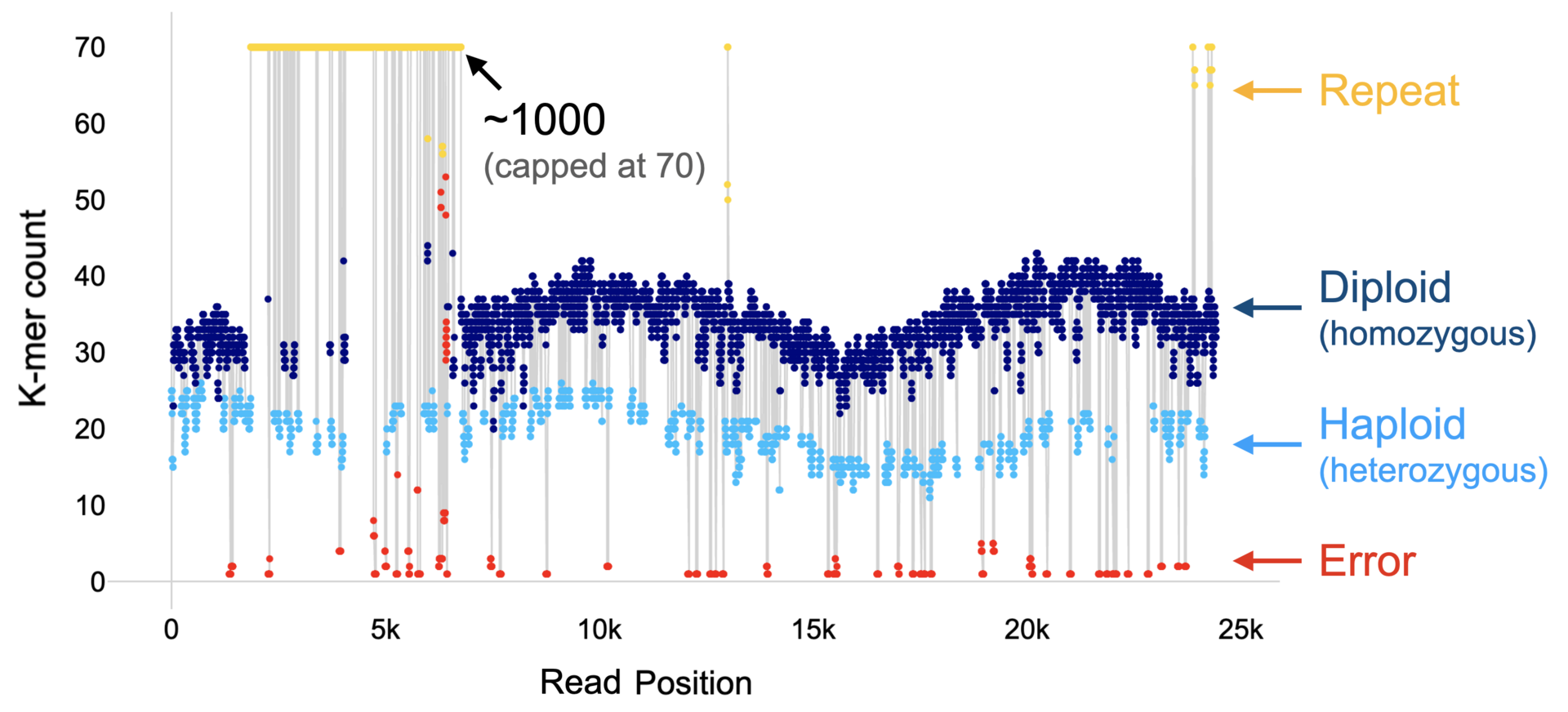
Figure 1: The k-mer profile of a read and its classification.
FastK produces directly the concept of a read profile (See Figure 1 above) and we have used the concept to very accurately determine the copy number of a k-mer in the underlying genome from the copy numbers in the read data set. Postdoc Yoshihiko Suzuki developed the algorithm and a software program callled ClassPro. Many analyses and inferences in the assembly of sequence data benefit from this increased classification accuracy in terms of efficiency and correctness. We are now exploring these possibilities. Several manuscripts are in preparation.
4. Publications
4.1 Journals
- Morishita, S., Ichikawa, K., Myers E.W. Finding long tandem repeats in long noisy reads. Bioinformatics 37 (5), 612-621
- Suzuki, Y., Myers, E.W., Morishita, S. Rapid and ongoing evolution of repetitive sequence structures in human centromeres. Science advances 6 (50), eabd9230
- Rhie, A., McCarthy, S.A., Fedrigo, O., ... Myers, E.W., Durbin, R. Phillippy, A.M., Jarvis, E.D. Towards complete and error-free genome assemblies of all vertebrate species. Nature 592 (7856), 737-746
4.2 Oral and Poster Presentations
- Myers, E.W. Counting, Profiling, and Classifying K-mers and Their Applications. BioDiversity Genomics 2021, talk, virtual meeting Oct. 2021.
- Suzuki, Y., Myers, E.W. ClassPro: K-mer Classification Based on Read Profiles. BioDiversity Genomics 2021, talk, virtual meeting Oct. 2021.