

FY2014 Annual Report
Physics and Biology Unit
Associate Professor Jonathan Miller
Abstract
The unit pursues biology of evolutionary genomics and informatics with physics-based tools and concepts. In FY2014 these activities included studies of sequence duplication, directed evolution, and mutual information between populations of neurons in vitro.
1. Staff
- Dr. Eddy Taillefer, Group Leader (through August 31, 2014).
- Dr. Kun Gao, Post-doctoral Scholar
- Dr. Zdenek Lajbner, JSPS Post-doctoral Scholar
- Dr. Reuven Pnini, Technician
- Mr. Quoc-Viet Ha, Technician
- Midori Tanahara, Research Administrator
2. Collaborations
- Theme: Neuroscience
- Type of collaboration: Joint research
- Researchers:
- Professor Gordon Arbuthnott, OIST
- Dr. Marianela Garcia Munoz (Arbuthnott Unit, OIST)
- Theme: Genome Evolution
- Type of collaboration: Joint research
- Researchers:
- Professor Damian Dowling, Monash University
- Florencia Camus, Monash University
3. Activities and Findings
3.1 Repeat length distributions in natural genomes.
Our unit's study of inter-genome and intra-genome sequence conservation has been comprehensively described in past annual reports. The algebraic distribution of duplicated sequence lengths for the non-repetitive [ i.e. repeat-masked ] natural genomes has been demonstrated by us and confirmed by the Arndt group at the Max Planck Institute for Evolutionary Genetics in Berlin, Germany, and both groups demonstrated models to account for the algebraic distribution. Among the important insights into the biology of sequence evolution that has emerged is that repetitive sequence does not conform to this distribution.
One of our principal conceptual and computational innovations in whole-genome characterization of sequence duplications is illustrated here by a popular quotation often misattributed to Gandhi, selected solely for its repetitive character:
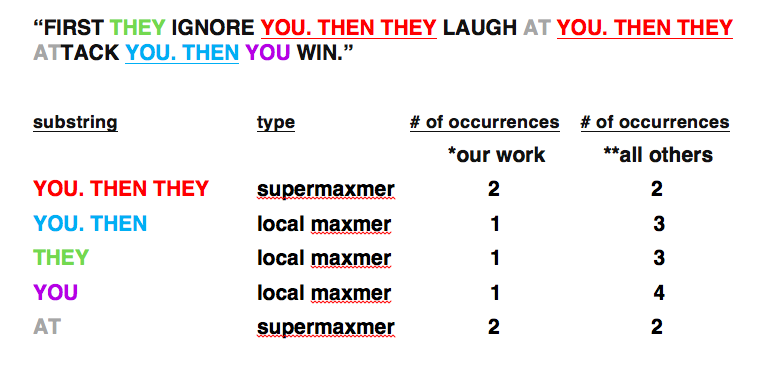
*Miller and co-workers; SEQANALYSIS [TM]. Observe that this classification of substrings counts each location in the genome no more than once.
**Arndt and co-workers [MA]; MUMMER and all other software of which we are aware that predates our work. Observe that this classification of substrings may count a single location in the genome multiple times. In this sense this counting is redundant, in effect integrating over longer substrings.
Subsequently many standard software packages, including MUMMER, were modified by their authors to better account for local maxmers in one way or another. User must take care with options on all versions of all software.
In most natural genome sequences, for long substrings, it is an empirical fact that supermaxmers dominate so that * and ** don’t differ that much; however, short substrings are another matter: ** and * diverge from one another strongly. The ** counting yields a total number of nucleotides that exceeds the length of the [ repeat-masked ] genome because in practice it counts each location in the genome multiple times.
The differences in counting quantitatively explain the discrepancies in length distribution for short substrings reported by Massip and Arndt [MA] and by Koroteev and Miller [KM]. The new length scale (“short” versus “long”) is reflected in natural sequence data and in the additional term that arises in the dynamical equations of KM and that is absent from MA. This additional term of KM accounts for annihilation of supermaxmers that does not conserve total substring length, because a large part of a substring can be destroyed.
The two ways of counting can be reconciled within an overarching framework by describing occurrences of a local maxmer as "nested [within a supermaxmer]" and occurrences of a local maxmer outside any supermaxmer as "non-nested." Nesting is thus a context dependent property. The suffix array based software, SEQANALYSIS, reports on a whole-genome scale the numbers, nesting, and locations of maxmers and their parents as a tool to study the evolution of duplicated sequence.
[MA] F. Massip and P. F. Arndt, “Neutral evolution of duplicated DNA: An evolutionary stick-breaking
process causes scale-invariant behavior,” Physical Review Letters, vol. 110, no. 14, pp. 148 101(1-5), April 5, 2013; e-publication date: April 2, 2013.
[KM] M.V. Koroteev, J. Miller, “Fragmentation dynamics of DNA sequence duplications,” submit/0689201; April 4, 2013. http://arxiv.org/abs/1304.1409; M.V. Koroteev and J.Miller, “Scale-free duplication dynamics: A model for ultraduplication,” Physical Review E, vol. 84, no. 6, pp. 061 919(1–10), Dec. 2011.
[TM] E. Taillefer, J. Miller, “Algebraic length distribution of sequence duplications in whole genomes,” in International Conference on Natural Computation, vol. 3, Shanghai, China, pp. 1454–1460. July 2011; E. Taillefer, J. Miller, “Exhaustive computation of exact sequence duplications in wholegenomes via super and local maximal repeats,” in International Conference on Computer Engineering and Bioinformatics, vol. 21, Cairo, Egypt, pp. 22–29. Oct. 2011; Taillefer E, Miller J. "Exhaustive computation of exact duplications via super and non-nested local maximal repeats," Journal of bioinformatics and computational biology. 12: 1350018, 2014. doi: 10.1142/S0219720013500182.
3.2 Directed evolution of mitochondrial genome.
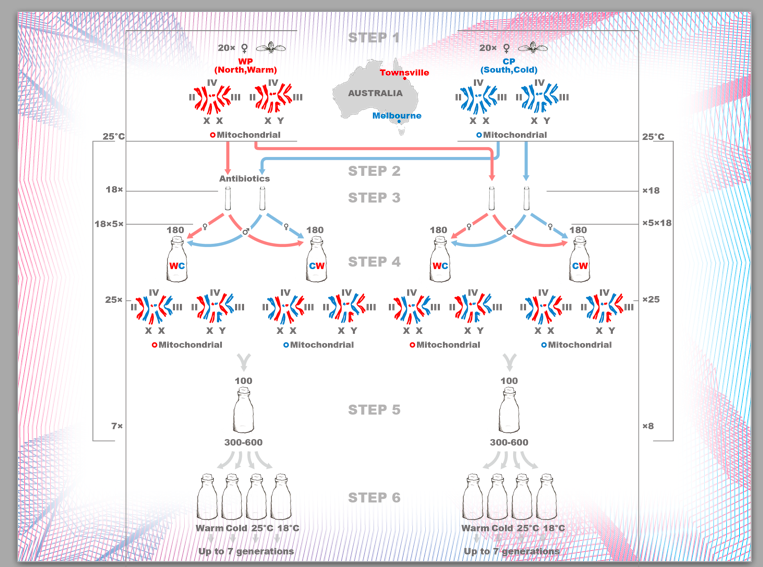
Dr. Zdneke Lajbner continued his characterization of the haplotypes of fruitflies that he bred in our laboratory under controlled thermal selection, in collaboration with the Dowling laboratory at Monash University. The original progenitor fly lines were obtained from the extremes of temperature on the Eastern coast of Australia.

His experimental protocol is indicated below:
3.3 "Facebook" for neurons.
In a challenging and technically difficult experiment, our collaborators in the Arbuthnott laboratory at OIST cultured populations of striatal neurons and of cortical neurons on opposite ends of a multi-electrode plate, and then removed a barrier between the two populations to allow them form connections with one another as they do in vivo. The acid test was to sever any connections by cutting across the middle of the plate with a knife. A photograph of a plate, with thirty electrodes in the top half (cortical cells) and thirty electrodes in the bottom half (striatal cells) is shown below; blue represents a recording of the potentials from the corresponding electrode as a function of time.
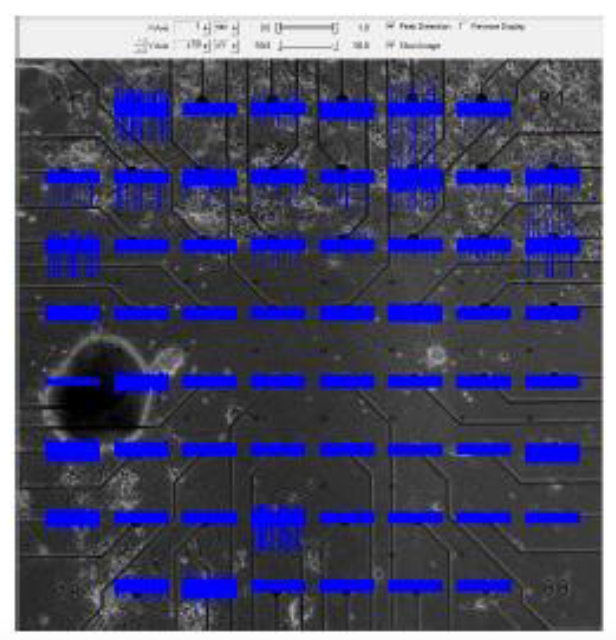
Our role, implemented by Dr. Reuven Pnini and Dr. Eddy Taillefer, was to establish whether these connections are active by studying the potentials recorded from the electrodes. They compute pairwise mutual information and correlation between the signals from the striatal and from the cortical populations. Active connections can be inferred only if signal appears after the connections have formed, but vanishes into the noise once the connections are severed. Pairwise correlation and mutual information among cortical electrode pairs is always strong, so that correlation or mutual information between a cortical electrode and a striatal electrode does not imply that neurons recorded by them are directly connected.
Some of the steps of Dr. Pnini's analysis of these data are indicated in the figures below.
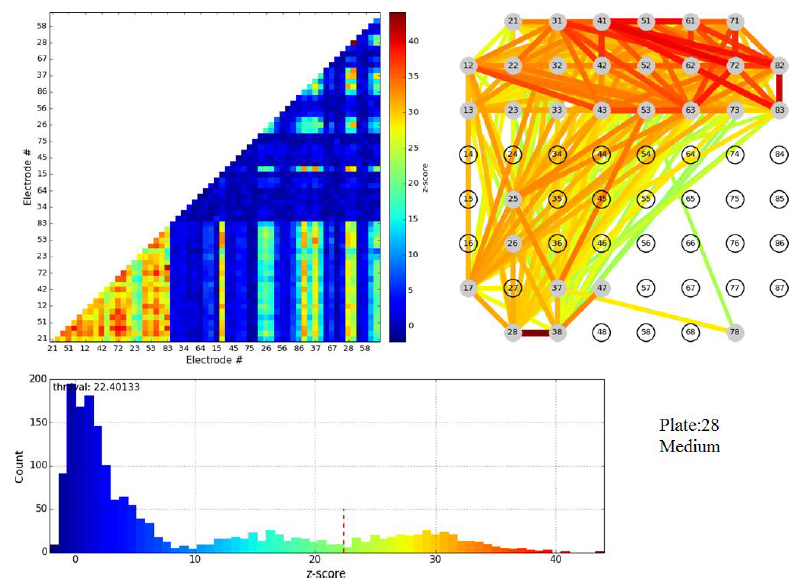
Figure 3.1: z-scores for pairwise electrode mutual information of a single plate shortly before cut. Top left: color map (the lower left corner corresponds to cortical-cortical electrode pairs). Top right: links between electrodes color coded to indicate z-score of mutual information in the linked electrode pair.
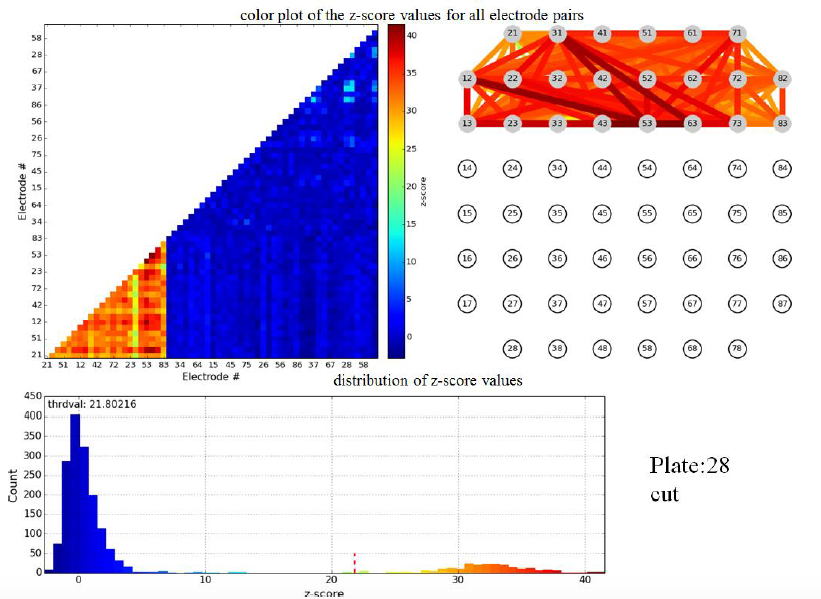
Figure 3.2: z-scores for pairwise electrode mutual information in same plate as figure 3.1, but directly after cut.
Evidently the cut wipes out the pairwise mutual information between cortical and striatal electrodes, validating the formation of active connections between these populations in vitro. We stress that the principal scientific advance described in this subsection of Activities and Findings is the experimental achievement of active connections in vitro between cortical and striatal neurons by the Arbuthnott lab. Nevertheless, Dr. Pnini and Dr. Taillefer overcame significant challenges in signal processing to develop and implement a straightforward, unassailable, and - just as important - simple computational pipeline to exhibit the activity of the neural connections from the multi-electrode recordings.
4. Publications
4.1 Journals
Gao K, Miller J. Human-chimpanzee alignment: ortholog exponentials and paralog power laws. Computational biology and chemistry. 53 Pt A: 59-70, 2014. doi:10.1016/j.compbiolchem.2014.08.010.
Taillefer E, Miller J. Exhaustive computation of exact duplications via super and non-nested local maximal repeats. Journal of bioinformatics and computational biology. 12: 1350018, 2014. doi: 10.1142/S0219720013500182
4.2 Books and other one-time publications
Nothing to report
4.3 Oral and Poster Presentations
Gao K, Miller J. Finding orthologs genomewide with NO alignment and NO repeatmasking. OIST, Okinawa. Feb 6 2015.
5. Intellectual Property Rights and Other Specific Achievements
Nothing to report
6. Meetings and Events
Nothing to report
7. Other
Nothing to report.