

FY2012 Annual Report
Open Biology Unit
Adjunct Professor Hiroaki Kitano

Abstract
Our unit aims to develop a novel software platform for systems biology and systems drug discovery that may fundamentally transform the way we study these fields.
Modern biology and medicine are fundamentally data- and evidence-driven, and researchers are fighting against the vastness and complexity of the data and the scattered nature of our knowledge. In order to obtain in-depth insights that may lead to new biological discoveries and medical implications, one must be able to properly access this data and knowledge and then integrate, analyse, and link them to practical solutions. This requires a new approach in science in the sense that it has to be open-ended, evolvable, community-based, and computationally supported.
Our unit is developing a methodology and software platform for open biology that entails the above features. This includes the Payao community-based pathway curation system, PhysioDesigner physiological modeling tool, drug-molecular network interaction prediction software, and a series of software packages that will be interoperable to The Garuda Platform. The Garuda Platform is a global alliance that we initiated to create a consistent, integrated user experience - a one-stop service style software platform. Such a platform will enable researchers to explore system-level characteristics of biological systems more effectively than before. At the same time, we are exploring a novel drug discovery approach based on computational systems biology, theory of biological robustness, and a concept of long-tail drugs.
1. Members
- Dr. Hiroaki Kitano, Projessor (adjunct)
- Dr. Yoshiyuki Asai, Group Leader
- Dr. Kun-Yi Hsin, Researcher
- Takeshi Abe, Technical Staff
- Kyota Kamioshi, Technical Staff
- Ken Kuwae, Technical Staff
- Midori Tanahara, Research Administrator
2. Collaborations
- Theme: Software platform for systems biology
- Type of collaboration: Research Collaboration
- Researchers:
- Dr. Samik Ghosh, Systems Biology Institute
- Ms. Yukiko Matsuoka, Systems Biology Institute
- Theme: Integration of Manchester text mining system to Payao
- Type of collaboration: Research Collaboration
- Researchers:
- Professor Sophia Ananiadou, Manchester University
- Theme: Open platform for multi-level modeling and simulation
- Type of collaboration: Joint research
- Researchers:
- Professor Taishin Nomura, Osaka University
- Professor Hideki Oka, RIKEN
- Professor Yoshihisa Kurachi, Osaka University
- Professor Kun-ichi Hagihara, Osaka University
- Dr. Masao Okita, Osaka University
- Professor Akira Amano, Ritsumeikan University
- Dr. Fumiyoshi Yamashita, Kyoto University
- Theme: Modeling and database with dynamic brain platform
- Type of collaboration: Joint research
- Researchers:
- Professor Hiroaki Wagatsuma, Kyushu Institute of Technology
- Professor Yoko Yamaguchi, RIKEN
- Neurosignal analysis
- Type of collaboration: Joint research
- Researcher:
- Professor Alessandro E. P. Villa, University of Lausanne
- Theme: 4D Visualization of simulation results
- Type of collaboration: Joint research
- Researchers:
- Dr. Ryo Haraguchi, National Cerebral and Cardiovascular Center
- Dr. Takashi Ashihara, Shiga University
- Theme: Cloud supporting simulation service; Flint K3
- Type of collaboration: Joint research
- Researchers:
- Dr. Nobukazu Yoshioka, National Institute of Informatics
- Shigetsohi Yokoyama, National Institute of Informatics
- Masaru Nagaku, National Institute of Informatics
3. Activities and Findings
3.1 Garuda Platform

One of the major focus of the unit is to launch and lead the Garuda Alliance that is designed to remedy various interoperability issues in systems biology and biomedical software tools and data resources. It aims at providing a coherent and comprehensive software, data, and knowledge platform for systems biology and biomedical research to maximize efficacy of software and computational oriented research while significantly benefitting end-users (typically bench biologists, bioinformatics person at the user site, and those who are in related industrial sectors).
One of the Garuda Alliance’s intentions is to provide a novel open innovation platform that may dramatically improves R&D productivity of basic biology research and pharmaceutical industry. We strived to accomplish these objectives by providing mechanisms for global distribution of computational, software, and knowledge base resources as a one-stop service portal, unified application program interface (Garuda Core API), consistent user experiences, and solid user supports.
Garuda workshops held in FY2012:
- Garuda Nine Workshop @ OIST Feb 19-22, 2013
- Garuda Eight Workshop @ ICSB Toronto Aug 18, 2012
3.2 Development of software for multilevel modeling: PhysioDesigner

PhysioDesigner is a software that supports to create computable models of physiological systems with spatiotemporal multiple levels. Models built on PhysioDesigner are written in PHML format, which is an XML based specification taking over from ISML. We have rebranded ISML to PHML in December, 2011. In FY2012, we have released three versions of PhysioDesigner, i.e. 1.0 beta1 (April, 2012), 1.0 beta2 (September, 2012), and 1.0 beta3 (January, 2013) at physiodesigner.org (Fig. 2).
Figure 2: A snapshot of the official website for PhysioDesigner.
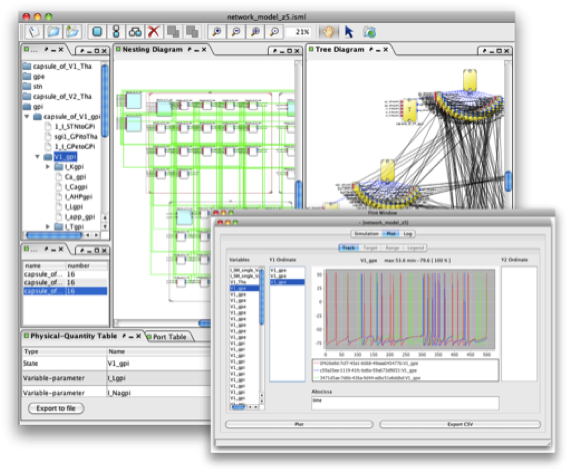
Figure 3: Snapshots of PhysioDesigner and Flint.
The major achievements on PhysioDesigner development in FY2012 were development of functions (1) to support of Garuda platform (http://www.garuda-alliance.org/), (2) to semi-automatically link edges among instances for large scale modeling, (3) to create a computational model as an aggregate of modules based on a 3D volume object such as a heart, (4) to visualize several medical image formats, such as NIfTI, analyze and so on.
We often find physiological structures composing of plenty of similar sub-structures, such as muscle tissue as an aggregate of many muscle cells and neural network consisting of many similar neurons. To model such a physiological systems, PhysioDesigner equips a template/instance framework to deal with those repetitive structure systematically. Multiple instances can be created according to the template. Instances are not simple ``copy" of a template. All properties of instances are inherited from the template, but each instance itself does not have concrete definitions of physical-quantities such as states and parameters. Since all instances follows the configuration of their template, functional edges connected to the template are considered to be connected all instances as well. This is convenient when all instances need to receive some information from other modules.
Template/Instance framework can be powerful method to create, for example, a whole organ model, because usually certain amount of cells of the same type are involved forming the organs, which can be modelled by instances. For such modeling, we also need to consider the morphological property. PhysioDesigner can utilise morphometric data with voxel-based volume representation and template/instances for creating such a model. A voxel-based volume model is an aggregate of volume elements, representing values on a regular grid in three dimensional space. By replacing each voxel by an instance and defining how an instance links with adjacent instances (in 6- or 26-directions), a computable model is created based on a voxel-based volume object (Fig. 4).
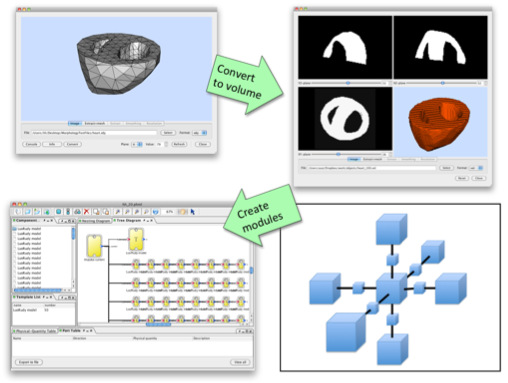
Fig. 4 Schema to create a model using template/instance framework based on a volume object.
3.3 Development of software for visualization of simulation results: PhysioVisualizer
Using PhysioDesigner, we can create multilevel large scale computable models of physiological systems. Flint can conduct the simulations of those models. What was missing was a visualizer of the simulation results. Especially as above mentioned, we now can create a model based on morphological data with template/instance framework, a visualizer which can map the simulated values onto the morphological objet was demanded. PhysioVisualizer deals with the morphological object and simulation data with the concept of the layers. Namely it can overlay several layers in one visualization (Fig. 5).
Although the basic functions were already implemented, PhysioVisualizer is still under development. It will be able to output a movie file as a result of the visualization of Flint simulation data with volume object data.
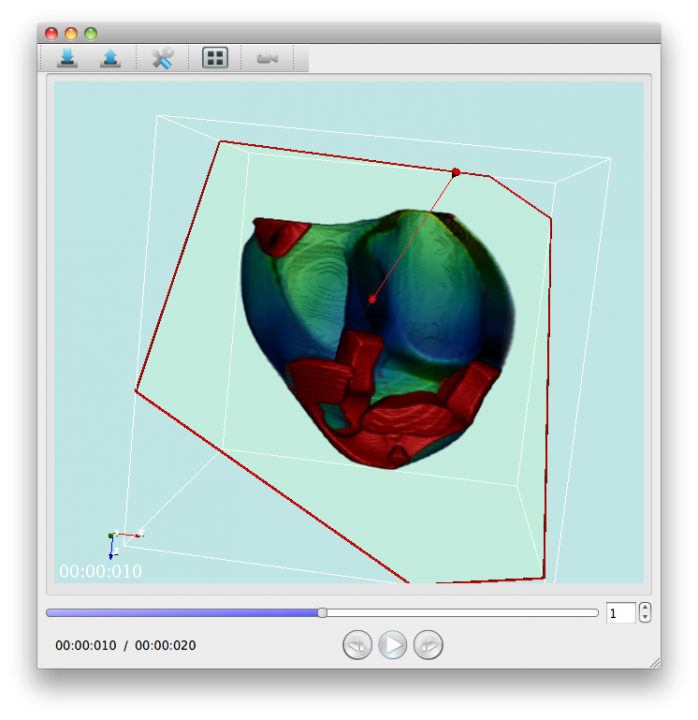
Fig. 5 A snapshot of PhysioVisualizer.
3.4 Development of software for simulations of multilevel physiological models: Flint

Flint is a simulator aiming to be capable of simulating a multilevel physiological models described in PHML/SBML. At the end of FY2012, we have released Flint 1.0 beta 3 for those who interested in running simulations of such models on their desktops. Now it comes with better performance and support of latest PHML features as well as useful functions such as rendering logarithmic scale graphs. We also have enhanced its interoperability with existing desktop and web applications for biology by supporting Garuda API complied with the Garuda alliance (http://www.garuda-alliance.org/).
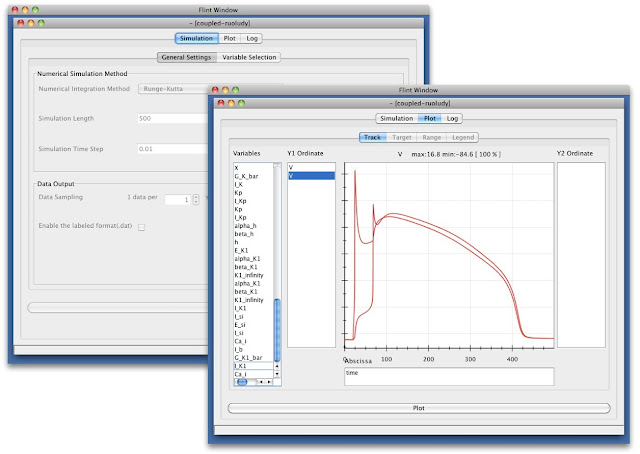
Fig. 6 Flint configuration and simulation windows.
3.5 Development of Flint K3 for cloud and HPC computing

Since the size of models is getting larger and larger nowadays, to enable Flint to work on high performance computers is demanded. We have been developing a Flint server, called Flint K3, working on computer clouds, so that users of PhysioDesigner can immediately send simulation jobs to high performance computing environment even if users do not have any accesses to high performance computers.
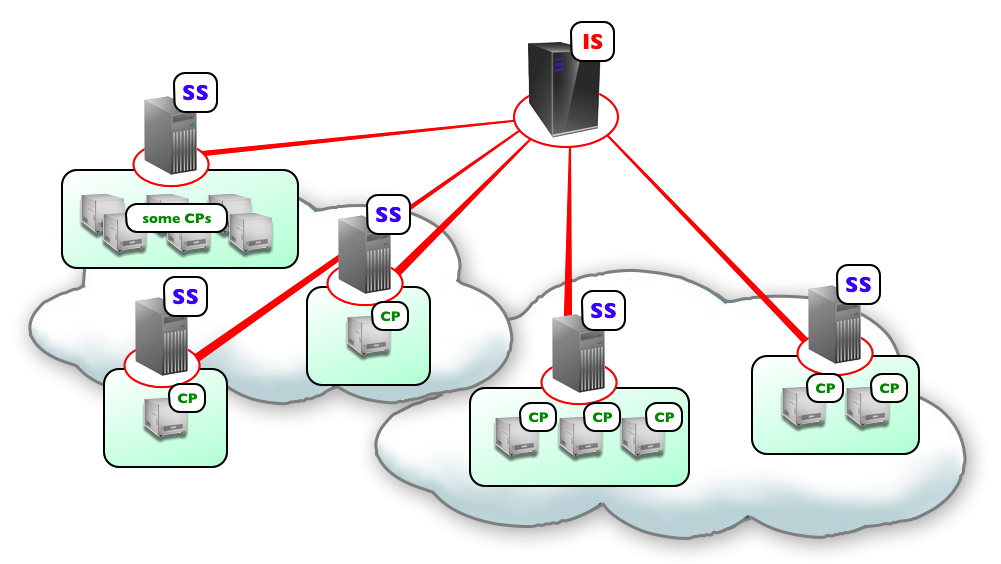
Fig. 7 Schema of Flint K3 architecture.
We have started K3 with ``edubaseCloud'' (http://edubase.jp/cloud) which is an open source based computer cloud for education of cloud engineering developed in National Institute of Informatics (NII). For development and preliminary test-run of K3, 64 cores on the cloud are assigned.
K3 is composed of two servers as shown in Fig. 7. One is an interface server (IFS), which receives job requests from users and manage the jobs. Simulation jobs are sent from IFS to simulation servers (SS) in the clouds. SSs evoke a computation program (CP) in every node assigned for the simulation. CPs perform numerical computation of the model in parallel having communications among them.
There are three ways for users to submit simulations jobs to Flint K3. One way is to visit the K3 IFS on a web browser. Users can upload models and configure simulation parameters for submitting simulation jobs at the site. The second way is to utilize a linkage between K3 and Model databases at open domain. There is PHML model database at Physiome.jp (http://www.physiome.jp/modeldb/) and SBML model database at several sites such as BioModels in EMBL-EBI (http://www.ebi.ac.uk/biomodels-main/). Users can provide a model ID defined in each database to K3 IFS. Then K3 IFS access to the database and download the model directly from the model database. Once such linkages among database and simulation server are built, users can easily check the dynamics of a model in database. The third way is to use the REST APIs implemented on IFS, so that applications such as PhysioDesigner can access to K3 directly, for example, to submit a simulation job and get the progress report of simulations.
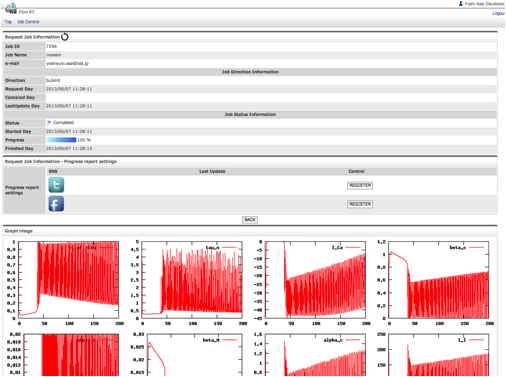
Fig. 8 A snapshot of FlintK3
3.6 Porting Flint to The K computer
We have ported Flint to the K computer (http://www.riken.go.jp/en/research/environment/kcomputer/) in order to take advantage of its computation power for simulating large-scale models. Combining inter-node parallel computation with better scheduling, Flint now calculates both ODE and DDE (Delay Differential Equation) faster, and it became possible to finish a simulation on a large model like heart cells within a feasible time span. The technique developed in this attempt is based on MPI so that it is portable up to recent HPC architecture, i.e., can be applied to porting it to any other HPC facility.
3.7 Development of a web and community based system for pathway publisher: iPathways+
Open Biology Unit and SBI has jointly developed Payao aiming of enabling a community to work on the same models of the cellular signal transduction simultaneously, insert tags to the specific parts of the model, exchange comments, record the discussions and eventually update the models accurately and concurrently.
Focusing on the feature of publishing the models, we have reengineered Payao based on JavaScript, and has started a sister service called iPathways+ at http://ipathways.org/plus/. iPathways+ does not equip interactive functions, instead, as a new feature, we added a function to enable users to embed a map in other web sites, as like google map. This feature has a big potential to expand collaborations with other database sites. Currently iPathways+ alpha version has been released. Besides that linkage between iPathways+ and existing social network service such as Facebook, Twitter and Google+ were enhanced, so that users can share models in iPathways+ over communities formed on the SNSs.
The development of iPathways+ has been promoted jointly by OIST and SBI.
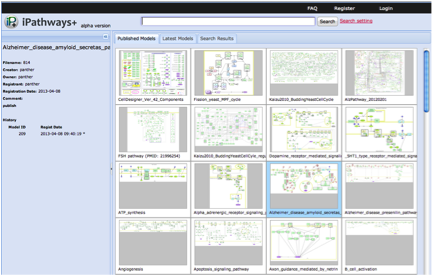
Fig. 9 Snapshots of iPathways+
3.8 High-precision in silico prediction approach for network pharmacology
Increased availability of bioinformatics resources is creating opportunities for the application of systems pharmacology to predict drug effects and toxicity led by multi-target interaction. Together with specialized bio- and chemo-informatics data, technologies including molecular interaction network description, structure-based drug design, high-throughput screening methods and statistical analysis help investigate the polypharmacology of a given drug/candidate. To achieve a comprehensive assessment of pharmacological effect in the early stages of drug discovery, we have been developing a high-precision in silico network-based screening approach for rapidly predicting the binding interactions between a given compound (e.g. lead/drug) against proteins involved in a complex molecular network (e.g. signaling networks). Novelty of the approach is composed of following points: 1) network-based screening, 2) incorporation of multiple molecular docking tools and SAR (structure–activity relationship), 3) application of machine learning systems and 4) open type innovation.
The machine learning systems that we have built are seen a great potential in improving docking simulation accuracy. Results from a series of validations and a case study show that our method possesses a competitive performance compared with state of the art (Fig. 10), as well as an adequate capability of identifying either primary or off-targets of numerous kinase inhibitors (Fig. 11). Such an in silico prediction approach has been implemented and deployed on OIST HPC together with a graphical interface for users not only bioinformaticist but also drugdevelopers who might not be familiar with a complex software operation.
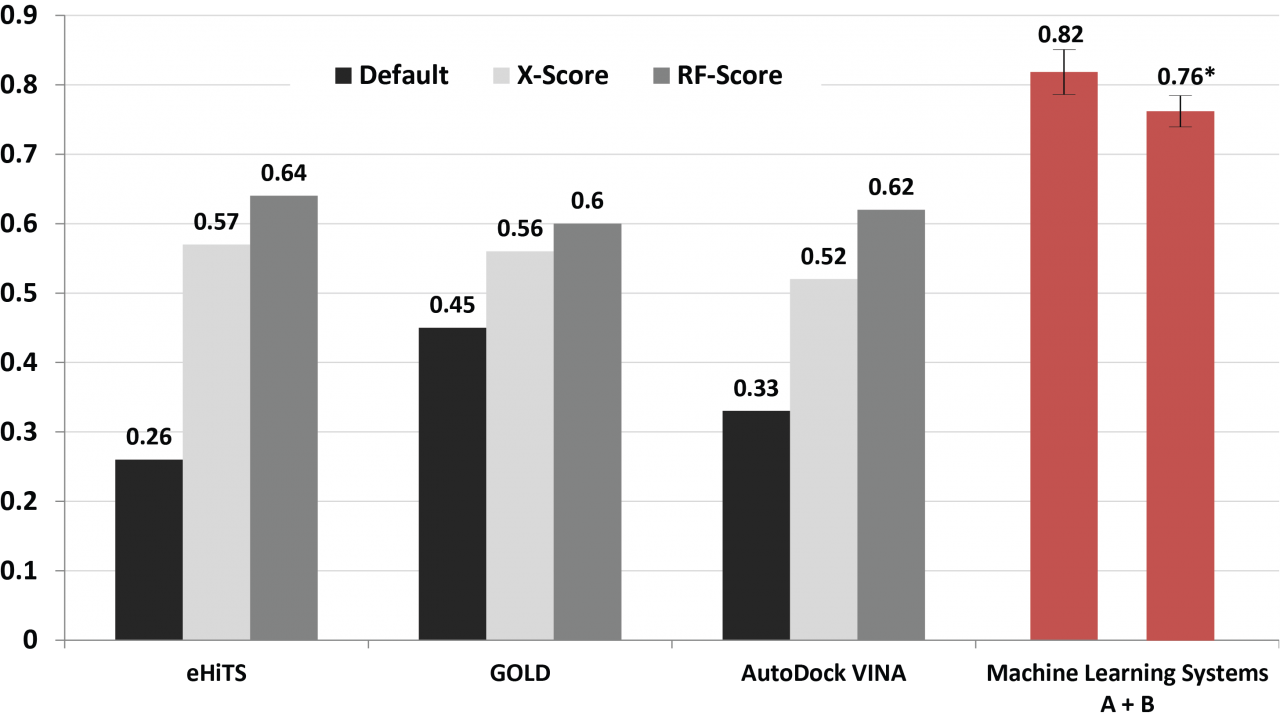
Fig. 10. Comparison of the prediction accuracy using different docking approaches.
Values are the correlations between the calculated scores and the corresponding experimental binding affinities. Black bars are the results using the default scoring functions equipped with the docking tools. Gray bars are those rescored by external scoring functions (i.e. X-Score and RF-Score) after docking. Red are performances of the machine learning systems A + B we developed using PDBbind version 2007 and 2012 datasets, respectively.
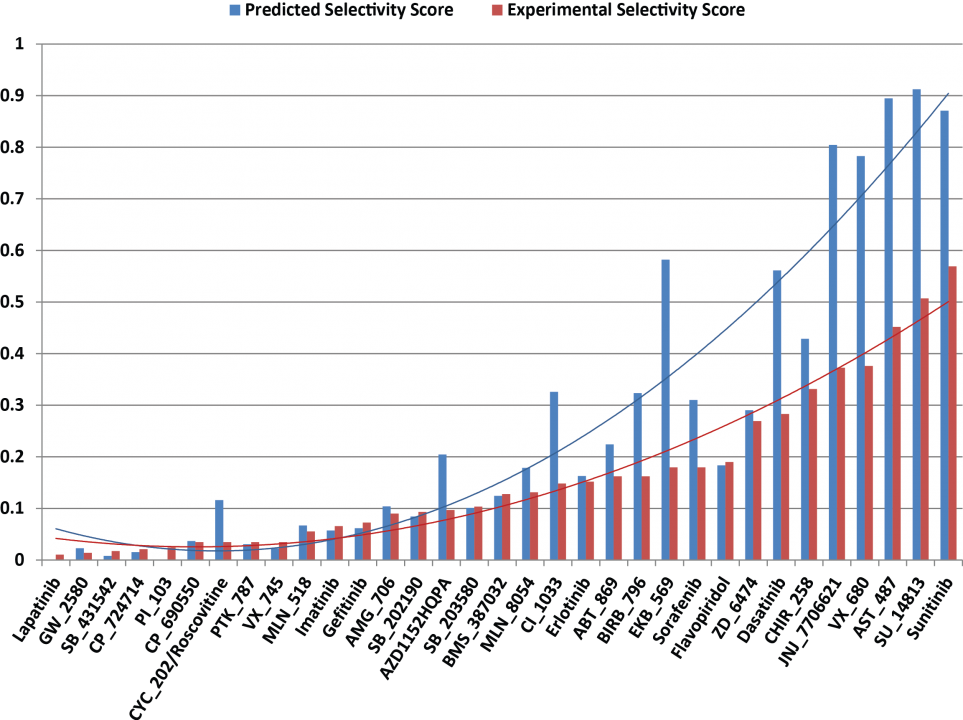
Fig. 11 Selectivity scores of the 33 kinase inhibitors against 139 different kinases.
A comparison is conducted by using the screening approach we developed (blue bars) and the referred bioassay results30 (red bars). The calculation of a predicted selectivity score is “S = number of docking score > 5.52 / number of kinases tested”, whereas the experimental selectivity scores is “S = number of binding affinity Kd < 3 µM / number of kinases tested”. A compound with a lower selectivity score indicates that it only actively interacts with a small number of target proteins, implying a lower potential of off-target effects.
4. Publications
4.1 Journals
- Carl-Fredrik Tiger, Falko Krause, Gunnar Cedersund, Robert Palmér, Edda Klipp, Stefan Hohmann, Hiroaki Kitano and Marcus Krantz*. A framework for mapping, visualisation and automatic model creation of signal-transduction networks. Molecular Systems Biology. 8: 578, April 24, 2012.
- Martijn P. van Iersel*, Alice C. Villeger, Tobias Czauderna, Sarah E. Boyd, Frank T. Bergmann, Augustin Luna, Emek Demir, Anatoly Sorokin, Ugur Dogrusoz, Yukiko Matsuoka, Akira Funahashi, Mirit I. Aladjem, Huaiyu Mi, Stuart L. Moodie, Hiroaki Kitano Nicolas Le Novere, and Falk Schreiber. Software support for SBGN maps: SBGN-ML and LibSBGN. Bioinformatics. 28 (15): 2016-21, 2012. doi: 10.1093/bioinformatics/bts270, published online May 10, 2012.
- Shoemaker, J. E*., Lopes, T. J., Ghosh, S., Matsuoka, Y., Kawaoka, Y., Kitano, H. CTen: a web-based platform for identifying enriched cell types from heterogeneous microarray data. BMC Genomics, 13 (1): 460, 2012.
- Shoemaker, J.; Fukuyama, S.; Eisfeld, A. J.; Muramoto, Y.; Watanabe, S.; Watanabe, T.; Matsuoka, Y.; Kitano, H.*; Kawaoka, Y. Integrated network analysis reveals a novel role for the cell cycle in 2009 pandemic influenza virus-induced inflammation in macaque lungs. BMC Systems Biology, 6:117, 2012.
- Martin H. Schaefer; Tiago J. S. Lopes, Nancy Mah, Jason E. Shoemaker, Yukiko Matsuoka, Jean-Fred Fontaine, Caroline Louis-Jeune, Amie J. Eisfeld, Gabriele Neumann, Carol Perez-Iratxeta, Yoshihiro Kawaoka, Hiroaki Kitano, Miguel A. Andrade-Navarro. Adding Protein Context to the Human Protein-Protein Interaction Network to Reveal Meaningful Interactions. PLOS Computational Biology. 9, 1, 2013.
- Koji Makanae; Reiko Kintaka; Takashi Makino; Hiroaki Kitano; and Hisao Moriya. Identification of dosage-sensitive genes in Saccharomyces cerevisiae using the genetic tug-of-war method. Genome Research. 23, 300-311, 2013.
4.2 Books and other one-time publications
- Kitano, H*. Cancer Systems Biology: A Robustness-Based Approach. HANDBOOK OF SYSTEMS BIOLOGY CONCEPTS AND INSIGHTS, Marian Walhout, et al.(ed.), 469-479, Academic Press, Dec.4, 2012.
- Samik Ghosh*, Yukiko Matsuoka, Yoshiyuki Asai, Hiroaki Kitano, Anshu Bhardwaj*, Vinod Scaria, Rohit Vashisht, Anup Shah, Anupam Kumar Mondal, Priti Vishnoi, Kumari Sonal, Akanksha Jain, Priyanka Priyadarshini, Kausik Bhattacharyya, Vikas Kumar, Anurag Passi, Pratibha Sharma, Samir Brahmachari. Software Platform for Metabolic Network Reconstruction of Mycobacterium tuberculosis. Systems Biology of Tuberculosis, Johnjoe McFadden, et al.(ed.), pp21-35, Springer, Dec. 6, 2012. . (ISBN 978-1-4614-4965-2)
- 北野宏明*. がんの多様性の数理. 実験医学2013年1月号, 31, 1, 45-52, Jan. 1, 2013.
4.3 Oral and Poster Presentations
- Yoshiyuki Asai, Takeshi Abe, Tatsuhide Okamoto, Hideki Oka, Taishin Nomura, Yoshihisa Kurachi, Hiroaki Kitano (2012) Multilevel modeling on PhysioDesigner. 第51回日本生体医工学会大会, 2012年5月10-12日(福岡)
- 吉川禎, 奥山倫弘, 置田真生, 浅井義之, 安部武志, 野村泰伸, 八木哲也, 萩原兼一. ``生体モデルにおける数式の類似性に着目したOpenMPによる並列シミュレーションの検討''. 第10回先進的計算基盤システムシンポジウム論文集(SACSIS 2012), 2012年5月16日 (招待講演)
- Kitano, H. Will engineering play the lead role in drug discovery in 2030? BioMelbourne breakfast, Cinema 1, Melbourne, Australia, June 5. (invited)
- Kitano, H. Software Platform for Systems Drug Discovery. Bio-IT World Asia Conference 2012, Marina Bay Sands, Singapore, June 8, 2012. (invited)
- Yoshiyuki Asai, Hiroaki Kitano (2012) Multilevel modeling of physiological systems and nervous systems using PhysioDesigner. IEICE Tech. Rep., vol. 112, no. 108, NC2012-12, pp. 93-95, Okinawa, Jun 2012. (invited)
- Kitano, H. Systems Drug Design and Garuda Software Platform. Network Biology SIG: On the Analysis and Visualization of Network in Biology (NetBio SIG), ISMB 2012, Long Beach Convention Center, USA, July 13, 2012. (invited)
- Kitano, H. HD-Physiology, Garuda, and Computational drug side-effects prediction. Talk at FDA, FDA, Silver Spring, USA, July 17, 2012. (invited) .
- Yoshiyuki Asai. PhysioDesigner: Current Status. 新学術領域研究「統合的多階層生体機能学領域の確立とその応用」平成24年度領域全体会議. 2012年7月22-24日(福岡)
- Yoshiyuki Asai, Takeshi Abe, Masao Okita, Tomohiro Okuyama, Nobukazu Yoshioka, Shigetsohi Yokoyama, Masaru Nagaku, Ken-ichi Hagihara, Hiroaki Kitano. (2012) Multilevel modeling of Physiological Systems and Simulation Platform: PhysioDesigner, Flint and Flint K3 service. Conf Proc 12th IEEE/IPSJ International Symposium on Applications and the Internet (SAINT 2012) July 27, 2012. pp.215-219
- Kitano, H. Biological Robustness. Seminar at University of Toronto, University of Toronto, Canada, Aug. 20, 2012. (invited)
- Kitano, H. Systems Biology powered by Artificial Intelligence. PRICAI-2012: 12th Pacific Rim International Conference on Artificial Intelligence (via skype), Pullman Hotel, Kuching, Malaysia, Sep. 7, 2012. (invited)
- Yoshiyuki Asai, Takeshi Abe, Hideki Oka, Masao Okita, Hagihara Ken-ichi, Yukiko Matsuoka, Samik Ghosh, Yoshihisa Kurachi, Hiroaki Kitano (2012) A platform for multilevel modeling of physiological systems: Template and instance framework for large scale models. 生体医工学シンポジウム2012,2012年9月7日(大阪)
- Hideki Oka, Ken-ichiro Iwasaki, Yoshiyuki Asai, Taishin Nomura, Yoko Yamaguchi. EEG Analysis in PhysioDesigner. NeuroInformatics 2012. September 10-12, 2012, Munich, Germany. p. 128.
- Yoshiyuki Asai, Alessandro E.P. Villa. Transmission of distributed temporal information on diverging/converging neural network and its implementation on a multilevel modeling platform. International conference on artificial neural networks. September 11-14, 2012, Lausanne, Switzerland.
- Kitano, H. VPH in industrial research. VPH 2012, Savoy Place, London, UK, Sep. 20, 2012. (invited)
- 北野宏明. システム創薬とGarudaプラットフォームの概要. BioJapan 2012, パシフィコ横浜, Oct. 11, 2012,
- Kitano, H. Systems biomedicine and their computational platforms. FOSBE 2012, Institute for Advanced Biosciences, Keio University, Oct. 21, 2012. (invited keynote)
- Yoshiyuki Asai, Hideki Oka, Alessandro E.P. Villa, Hiroaki Kitano. Multilevel modeling platform and its application for modeling in neuroscience. 2012 International Symposium on Nonlinear Theory and its Applications (NOLTA). October 22-26, 2012, Palma, Majorca, Spain.
- Yoshiyuki Asai, Takeshi Abe, Hideki Oka, Yoshiyuki Kido, Li Li, Taishin Nomuran, Yoshihisa Kurachi, Hiroaki Kitano. Multilevel Modeling and Simulation of Physiological Systems on PhysioDesigner. INCF Japan Node International Symposium. ADVANCES IN NEUROINFORMATICS 2012. 2012年10月30日
- 北野宏明. システムバイオロジー概論. 第6回KASTシステムバイオロジー講座, かながわサイエンスパーク, Nov. 2, 2012. (invited)
- Kitano, H. Systems Toxicology. An invited talk at DSTO, Defence Science and Technology Organisation (DSTO), Department of Defence, Australian Government, Melbourne, Australia, Dec. 4, 2012. (invited) .
- 北野宏明. Data-Driven Network-Based Biomarker Discovery. 第35会 日本分子生物学会年会 バイオテクノロジーセミナー, 福岡国際会議場, Dec. 12, 2012. (invited)
- Yoshiyuki Asai. PhysioDesigner 1.0 beta3. 新学術領域研究「統合的多階層生体機能学領域の確立とその応用」平成24年度領域全体会議. 2013年1月16-18日(福岡)
- Yoshiyuki Asai, Alessandro E.P. Villa. スパイクパターンに基づいたフィードフォワードニュラールネットワーク内の神経活動伝達に関する考察.信学技報 (Technical Report of IEICE) CAS2012-86, pp.115-119
- 北野宏明. システムズバイオロジーのための京コンピュータへの期待. 第4回スーパーコンコンピュータ「京」と創薬・医療の産学連携セミナー -HPCI計算生命科学推進プログラム-, フクラシア東京ステーション, Jan. 25, 2013. (invited) )
- Yoshiyuki Asai. Demonstration of PhysioDesigner 1.0 beta3 and Flint with Garuda.Garuda Nine Workshop. Okinawa, Japan. Feb. 19-22, 2012.
- Yoshiyuki Asai, Hiroaki Wagatsuma. J-Node PFs + Garuda, PhysioDesigner and Flint. Dynamic Brain Platform Meeting. 2013年2月27日(北九州)
- Kitano, H. Systems Drug Design and Software Platform for drug efficacy and adverse effects prediction. FDA Workshop: Systems Pharmacology for the Prediction of Tyrosine Kinase Inhibitor non-QT Cardiotoxicity, FDA White Oak Campus, USA, Feb. 28, 2013. (invited)
- Yoshiyuki Asai, Hiroaki Wagatsuma. Garuda, PhysioDesigner and Flint with J-Node PFs. NIJC meeting. 2013年3月6日(和光)
- Tomohiro Okuyama, Masao Okita, Takeshi Abe, Yoshiyuki Asai, Taishin Nomura, Hiroaki Kitano, and Kenichi Hagihara. Accelerating General and Heterogeneous Biophysical Simulations Using the GPU. In Poster in the 4th GPU Technology Conference (GTC 2013), San Jose, CA, USA, (2013-03).
5. Intellectual Property Rights and Other Specific Achievements
- 特許/特願2012-134261
- 特許/特願2012-245326
- US仮出願61671049
6. Meetings and Events
6.1 HD-Physiology 6th plenery meeting and young researchers workshop (externally organized)
- Date: January 16-17, 2012
- Venue: OIST Campus Seminar Room B250/C209
- Co-organizers: Osaka University ad OIST (sponcered by KAKENHI MEXT Innovative Areas HD-Physiology Project)
6.2 Garuda Nine Workshop
- Date: February 19-22, 2013
- Venue: OIST Seaside House
- Co-organizers: OIST and SBI
