

Research at OIST - Ester Metathesis and Transfer Hydrogenation of Esters
Ester Metathesis
While I was developing the Wittig coupling chemistry of alcohols, something that was suggested by David (Milstein, the postdoc boss https://www.weizmann.ac.il/Organic_Chemistry/milstein/) was to try the reaction of an ylide together with an ester in the presence of the dehydrogenation catalyst. The thought was to maybe get some sort of catalytic Tebbe’s reagent, but it was a long-shot and unrealistic. The result that I saw was unexpected, and also not that interesting from the point of view of the Wittig and Tebbe reagent chemistry. There was majority olefin product formed with the alkoxy part of the ester, but also a little bit with the acyl part of the ester. The reaction was not so selective when using unsymmetrical esters and even if using a symmetrical ester like ethyl acetate the yield was lower than if just using pure ethanol instead. Interestingly, when using an unsymmetrical ester like ethyl hexanoate, I found a little bit of hexyl acetate and hexyl hexanoate in the GC trace.
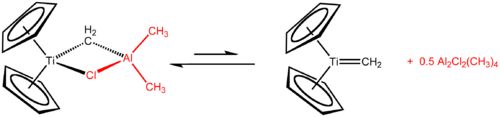
Tebbe reagent picture taken from Wikipedia

The real usefuleness of the Tebbe reagent is probably due to the selective alkylation of the acyl part of the ester and the creation of vinyl ethers thereby
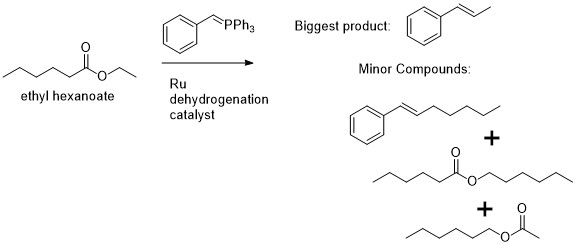
Some unexpected products that are observed in the GC trace when reacting an ylide and an ester in the presence of a Ru dehydrogenation catalyst
When starting to work in the lab at OIST, I decided to revisit this interesting result and try the reaction of an ester in the absence of ylide, to see if I could get a similar type of ester scrambling. I assumed at the time that the ester formed due to some decomposition that made it possible to generate the Ru dihydride complex, which is an intermediate often found by NMR in the reaction mixtures and is believed to be the resting state of the catalyst. Once this was formed and there was enough of a reservoir providing hydrogen, alcohols could be made from the ester by the catalyst and immediately dehydrogenated back to the ester, scrambling it along the way. All the previous reports of ester hydrogenation required at least 5 atmospheres of hydrogen pressure to generate the associated alcohols, so it seemed unlikely that I would see such a metathesis process here, but the small peaks that I saw in the GC/MS traces during the Wittig project gave some hope.

Various catalysts used in the literature that have good activity in ester hydrogenation but require pressure of hydrogen. In this work, the Kuriyama 2012 (PNP), Gusev 2012 (PNN), and Gusev 2013 (SNS) were used with the latter being the most active, and only the first and third being active in pure metathesis chemistry
At first in order to have enough of a hydrogen reservoir and not decompose the ester (at the time I thought that maybe the catalyst was just forming an olefin by dehydrogenating next to the acyl moiety), I added about 100% of ethanol to ethyl hexanoate and threw in 1 mol% catalyst and 2-3mol% of KOtBu in deuterated benzene. Thus, it was possible to heat the reaction and follow it by NMR. The catalyst was the commercially available Takasago MACHO catalyst. Now that I was really not at the Milstein group, I didn’t feel like using the Milstein catalyst since it (the bipyridyl one) is more difficult to make. And MACHO you just make an order online, while the Milstein bipyridyl one used for the Wittig coupling was not commercially available (I have made lots of the ligand recently so it’s not hard, but it’s just busy work).
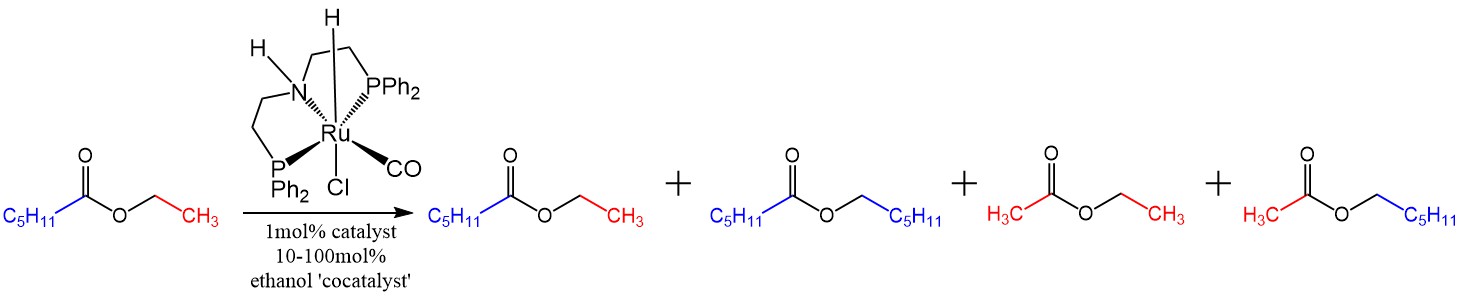
Metathesis of ethyl hexanoate with ethanol as an ersatz hydrogen source
The logic worked, and I did see some hexyl acetate, hexyl hexanoate, and ethyl acetate along with the starting material. At the same time, I also saw quite a bit of hexanol. This was not surprising since the extra ethanol that I threw in at the beginning was participating in the reaction and if it formed the acyl part of some ester, some of the ester that was there initially had to end up as an alcohol. Still, these results were encouraging because just right there they showed that it is not necessary to have much of a pressure of hydrogen to hydrogenate an ester (maybe less than 1 atm). So the next thing to do was to lower the amount of the hydrogen reservoir (ethanol) in order to avoid hexanol production, and have as much pure ester scrambling as possible instead. The reaction proved possible to do even at 50% of ethanol to the ester. Then it proved possible to do at 10% ethanol to the ester, with very little hexanol byproduct formed and lots of ester scrambling instead. Then I did the control experiment with absolutely no ethanol added at all, and the reaction still worked. That was surprising. Why would it work in the absence of any hydrogen source at all?
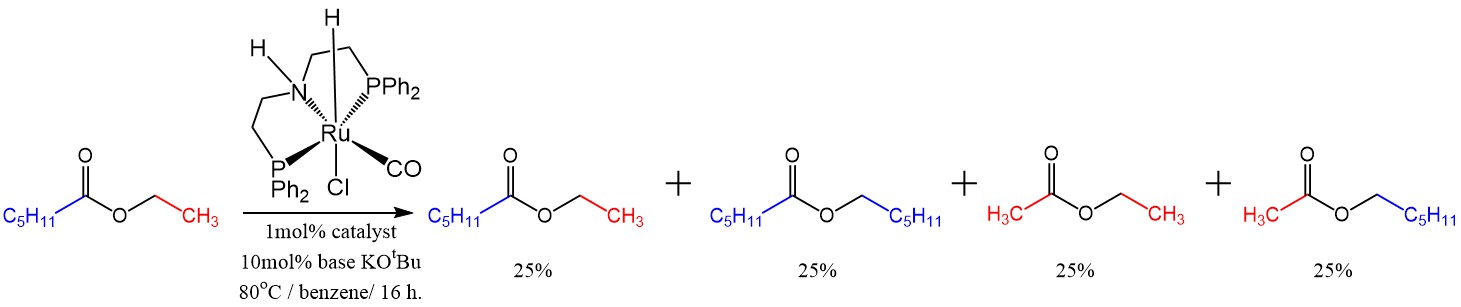
Metathesis with no hydrogen source at all. The ratios at the end were very close to 25.0 percent
Now it could get the hydrogen it needed by decomposing some of the ester and making a bit of olefin, but I didn’t even see traces of olefinic protons in the NMR. What was more important, was that substrates that did not have the option of making the olefin were also reactive in the scrambling reaction. A substrate such as tolyl benzoate for example. It meant that the catalyst was scrambling the ester all on its own without the need for extra hydrogen. By this time some other catalysts became available in the lab as shipments arrived and the glove box was set up, so I tested a few more commercially available catalysts. The MACHO one took a really long time to react and I was starting to use 5mol% to get quick ester metathesis (statistical distribution of all four possible esters), after one day. The Milstein catalyst that is commercially available is slightly different from the one I used in the Wittig coupling chemistry, but it was also a disappointment. There were two Gusev catalysts available commercially from Aldrich. One PNN pincer one and an SNS one that proved to be very unusual in that in had no phosphine donor, and was also reported to be much more active than MACHO in the hydrogenation of esters. The first PNN based one was completely inactive in the scrambling/metathesis of ethyl hexanoate, but the SNS based one proved to be more active than MACHO in metathesis actually. I could now get away with 1mol% and get statistical scrambling after 1 day at 80oC.
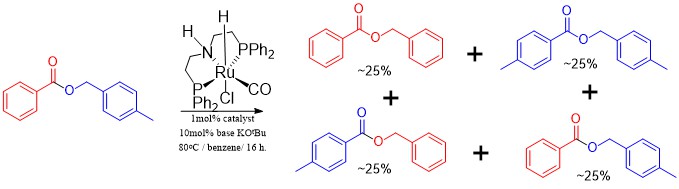
Metathesis of tolyl benzoate, which cannot be dehydrogenated to form an olefin
The catalysts that were active to some degree in ester metathesis all had one thing in common however. They were extremely sensitive to the type of substrate. Only hydrocarbon esters could react. If there was a heteroatom such as a halide, the reaction didn’t occur. Also, the reaction didn’t happen if the substrate was a methyl ester such as methyl hexanoate or methyl benzoate (or was a formate). It just set there in the NMR tube even if the catalyst loading was 5 mol%.
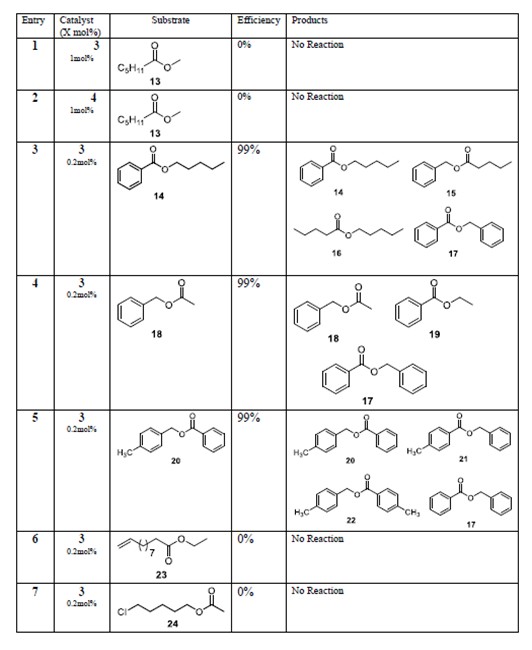
The unfortunately limited scope of the pure metathesis reaction
Of course, esters are not the same as olefins (of the more famous olefin metathesis fame). One of the more obvious differences is that the unsymmetrical olefins that can arise from the scrambling of 2 octene for example, will both be 2 octene, the same molecule (there are some finer details such as E/Z isomerism, but most catalysts will be selective for E olefins), with only the symmetrical products such as 2 butene and 6 dodecene being different. For an ester, the unsymmetrical products of the scrambling of ethyl hexanoate will be different however, unlike the case of the olefin. We should get an equal ratio of ethyl hexanoate and hexyl acetate, which have different physical properties. That means that the energies of the two products are slightly different, and we should expect a slight difference in their ratio. Now, for a simple carbon chain ester, the differences between the two possible positions of the acyl group are not going to be that significant, but for an ester such as ethyl benzoate for example, having the acyl group conjugated to the aromatic ring, will make a significant difference. I did a quick DFT calculation and sure enough, ethyl benzoate has a Gibbs free energy that is about ~2kcal/mol less than benzyl acetate. Taking the symmetrical esters ethyl acetate and hexyl hexanoate, adding their energies and dividing by two to compare them directly, we also get a value of ~1kcal/mol less than benzyl acetate.
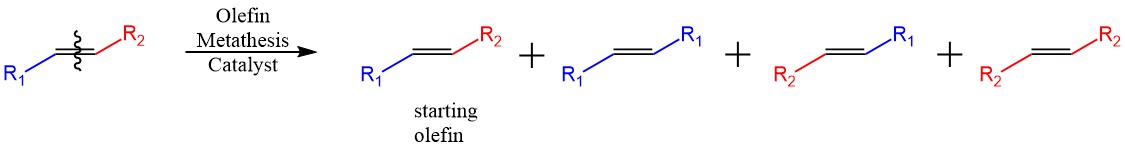

Ester and olefin metathesis comparted. The first and the third olefin are the same compound, but the first and third ester will be different.
This means that if we take benzyl acetate and throw in our SNS Gusev catalysis, to which by now I started referring to as the ‘metathesis catalyst’, then we should get more of the other products, depending on the time that we leave them to scramble. The number of TON required to reach statistical equilibrium gets more and more the closer you get to that equilibrium point, and it’s exacerbated by a low energy difference between the starting material and the reactants, but nevertheless we should see this effect after a while. Indeed, after 1 day I did see significantly more ethyl benzoate than the starting material (about 7:3) I believe, but I decided to stop it right there since waiting for something like a week to get a 9:1 ratio, was not really necessary for my purposes. Another thing that we could look at in this reaction was the rate of appearance of the products. Interestingly, at the beginning only the symmetrical esters ethyl acetate and benzyl benzoate appeared, even though they are ~1kcal/mol higher in energy than ethyl benzoate. It took a very long time for the actual most stable product to start appearing. After one day it was the major component, but this was not reflected by the initial value at all. The simplified mechanism that we came up with to explain it, was that the catalyst was involved in directly flipping the acyl and the alkoxy group of the ester in the first step, and the released ethoxy group would lead to the generation of a symmetrical ester by reacting with the benzyl acetate pool available in the reaction flask. The other partner, or benzoxy, would take a benzoate back from the catalyst to form the other symmetrical partner. This means that the rate of appearance of the symmetrical esters would be the same, and would slow down quickly after some time, as eventually when a big enough pool of them built up, the ethoxy could react with a free benzyl benzoate to form the most stable product, that we indeed do see slowly appear after a long enough reaction time. The quantitative measurement is a bit complicated by the fact that at 60oC the ethyl acetate tends to accumulate on the upper walls of the NMR tube to a small degree, but it’s fine for an accurate enough approximation. At least this possible mechanism (see the scheme below) explains the kinetics pretty well.
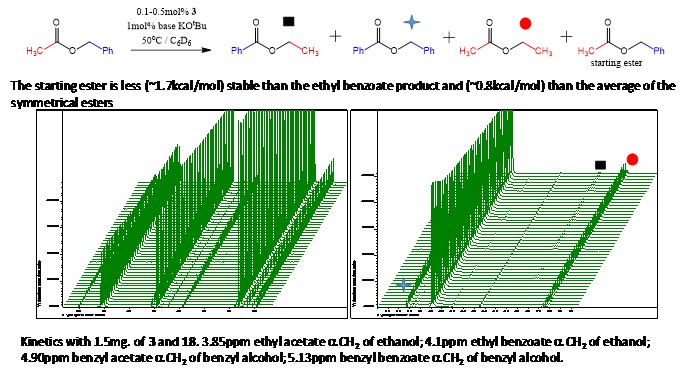
How the NMR kinetics experiment looks in real (fourier transformed time domain) life
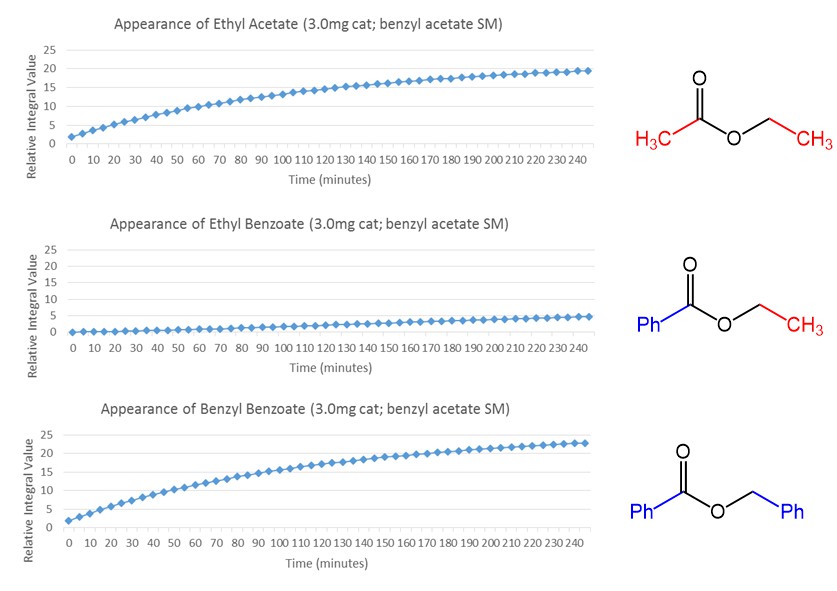
Data for the peaks plotted in order to compare concentration of products data

Possible mechanisms. The upper pathway is active in the absence of any hydrogen source and the second pathway becomes active when there is a hydrogen source present, explaining our transfer hydrogenation activity that is described later
It also explains why the catalyst is really sensitive to functional groups. We have a Ru center somehow holding on to an acyl and this could be a very reactive species. And no source of hydrogen anywhere around. So basically only hydrocarbons that could not generate methanol (and thus CO which would poison the catalyst) would work. At the time, I didn’t care too much because I thought it was a great result. This was true metathesis. Wasn’t this a great result that was just as good as olefin metathesis, but here we get scrambling around a completely new functional group? I was telling this to everyone and though it would get into a good journal. I have to admit that at the time I harbored delusions of even publishing it in Science. I tried to keep them as secret as possible because always at the back of your head there is a though that you will really embarrass yourself if you say that out loud (smart thinking in retrospect), but I might have told a few people…
Eventually, I talked to a few professors who told me that no, what I had was not that interesting beyond a minor curiosity.
“It’s a completely new reaction! You just take an unsymmetrical ester and the catalyst turns the acyl group into an alcohol, and the alcohol into the acyl. You get four products out in a statistical ratio just like in olefin metathesis!”
“Yes, but olefin metathesis is actually useful because you get one product out. You can make it selective.”
“No, it’s not selective! That’s the whole point of it. It’s a statistical scrambling. But it’s selective in that if they take 1-octene, they will eventually make ethylene and tetradecane. And ethylene is a gas, so it just flies away and you only have tetradecene left over.”
“So you see, it’s selective for tetradecene.”
“But the reaction itself is not selective! It’s being driven by equilibrium when you remove one of the products! When olefin metathesis was first reported, they weren’t reporting any selectivity, it was just an interesting reaction that scrambled olefins.”
“First of all, I’m not sure that true, but can you show selectivity towards one of your esters?”
“Well, if I use benzyl acetate, I get slightly more ethyl benzoate because it’s more stable, but it will never reach 100% of that product. At best maybe close to 50% after a really long reaction time since ethyl acetate and benzyl benzoate are close in energy…”
“Look, it’s just not practical. With olefin metathesis you can make polymers and it’s tolerant to functional groups. It’s used in the pharma industry in the synthesis of some drugs.”
“There are polyesters you know. The reason they can make polymers from something like cyclooctene is because you form ethylene and it flies away so they just force the equilibrium. At its base, it’s not a selective reaction. They are using that Le Chatelier trick. If I had something like that I could get selectivity as well, but for me methoxy just doesn’t work with these catalysts and with ethoxy I get ethyl acetate, which has a high boiling point. If I start refluxing stuff just to drive off the ethyl acetate, it’ll be a mess as it condenses back, and the catalyst is really sensitive, so an open reflux system is bad for it as something can get in and kill it. And you know what, I bet the first reported olefin metathesis was not really tolerant of functional groups either.”
“Is that true from the first reports? I’m not sure you checked. Can your catalyst make a polyester?”
“Well, polyester is made from a lactone, but it will just polymerize on its own with some base. There are reports of a completely new complex that is unusual, or an unsual transformation that make it into JACS. When I was a graduate student someone reported the first ever transfer hydrogenation of alkanes with a Pt catalyst. They only had 1.3 turnovers and it was a bit iffy if maybe it was not some artifact, but because it was the first ever reaction, it got into JACS.”
“I think your reaction is interesting, but these days you have to show some use. It might get into a minor journal on its own.”
Of course, the tendency after receiving such negative news about a reaction you care a lot about, is to be defensive. But, I learned only too well during my postdoc that you can get stuck thinking your chemistry is really good and find that it doesn’t seem that way to the outside world. This is not only true when you’re developing a new smart phone that makes 3D holograms that it turns out nobody wants to buy because you didn’t care to do your market research and users care more about battery life, it’s also very true for trying to publish in JACS or Angewandte. Here it’s a bit different since after all it’s chemistry and objectively you can be right. You could also be wrong of course and this is much more likely if you’re just one person disagreeing with a lot of other chemists. Ultimately, if you’re going against the general consensus of what type of chemistry is interesting and worthwhile to other chemists, you’re not going to be publishing in those journals. You’ve got to do your market research and you can’t get mad at the customer if they tell you something you didn’t want to hear (complaining about the customer at the bar on a Friday evening is a whole different matter of course).
Transfer Hydrogenation of Esters
So back to the drawing board, with my now useless and much despised ester metathesis reaction. The drawing board was basically in my head and it said: “didn’t you have that set of reactions when you were trying to optimize this earlier, before you did the control reaction? Something like, ‘So the next thing to do was to lower the amount of the hydrogen reservoir (ethanol) in order to avoid hexanol production and have as much pure ester scrambling as possible instead.’?” Right. I was getting hexanol byproduct if there was a hydrogen source such as ethanol, that was taking the acyl group into itself and forming ethyl acetate. What if I had lots of ethanol? Would I just be able to form hexanol in some sort of transfer hydrogenation of esters by a new mechanism?
Reaction that made me think that transfer hydrogenation of esters would be possible
In fact, it turned out that transfer hydrogenation of esters does not occur readily by any mechanism. There was a review on transfer hydrogenation that just came out, called ‘The Golden Age of Transfer Hydrogenation’, but esters were not mentioned. And there was a paper that came out a little bit before which claimed a first report of transfer hydrogenation. But at best they could get 9 TONs with an activated secondary alkoxy and really high catalyst loading. They used isopropanol as a hydrogen source. That means that acetone is formed as the acyl group sink to get the alcohol from the ester. When thinking about that a bit, I assumed that the energy of getting a ketone from an ester and forming a primary alcohol from a secondary one must be uphill. If I’m using ethanol and getting ethyl acetate, I’m exchanging one type of ester for another. I’ll be energy neutral or at worst off by a couple of kcals. I should be able to overwhelm that energy difference with enough ethanol. I tried it with 10 equivalents of ethanol to ethyl hexanoate, and it worked. I the NMR and the GC, I saw a whole bunch of free hexanol. Some of it was tied up in hexyl acetate or hexyl hexanoate, but the majority was definitely hexanol.
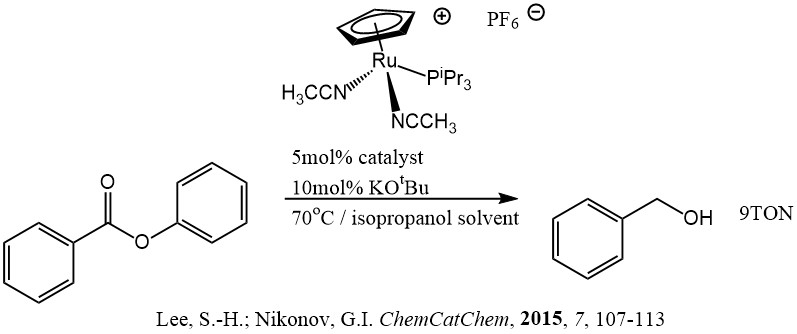
The only previous report of transfer hydrogenation of esters came out in the previous year. The result is far from practical as it is, but it shows that it may be possible
At this time I convinced one of Julia’s postdocs, Abhishek Dubey, to help me out in his free time by saying that the project will only take a little bit (our usual trick in the trade) and he can quickly do it on the side without affecting his work in the Khusnutdinova group if she was okay with it. It would lead to a very good publication on the first ever ‘(practical) Transfer Hydrogenation of Esters’ that would be published in some great journal and would be good for his career, etc…
Abhi optimized the reaction and made a few calibration curves for the GC/FID. We found out that it was best to do the reaction in toluene, but to use 20 equivalents of ethanol. Any more and it was too much for the catalyst, and it started to really slow down. 10 equivalents meant that the theoretical yield of hexanol was limited to ~90% and practically we could expect to get ~80%. After the success with ethyl hexanoate, we tentatively tried it out with other substrates that failed in ester metathesis. And things like methyl hexanoate and even esters with functional groups just started to work. I assumed it was because in the presence of a hydrogen source, the mechanism was different, and we can get that trans dihydride complex. The assumed mechanism is something that I envisioned from the beginning where we have the trans dihydride formed from the ethanol and it hydrogenates the target ester.
The functional group tolerance includes anything that won’t participate in the traditional amine and alcohol coupling chemistry. So primary and secondary amines are out, but tertiary amines are well tolerated. Pyridine rings are okay as well, as they don’t bind that strongly to the ruthenium. We also tried to hydrogenate some fatty acid esters and even though there was some indication that it worked, the yields of the long chain alcohols were not that high. This was actually mostly caused by solubility I believe, since we could see white globs of that fatty acid ester bobbing inside the hot toluene/ethanol reaction flask. Something to improve in the future.
![]()
Table of substrates that are active in transfer hydrogenation (definitely not an exhaustive list). Trilinoleate was disappointing, but this is likely due to solubility issues
Anyways, by now I finally had something practical and the feedback mill was telling me that I had a shot with JACS as long as I could figure out the mechanism. At this point I just said enough is enough, since plenty of people publish with preliminary mechanism when the reaction is great enough and here we have two completely new reactions, with one of them being sort of practical. Of course, we’ll ignore the fact that even 90% yields mean that you might still be better off with hydrogen gas pressure instead, it’s the concept of the first ever transfer hydrogenation of esters that matters.
So I submitted the paper and it got rejected after review due to having a crazy and not very believable mechanism. Now to be fair, the first mechanism that I drew did have a Ru formate species (that does exist in the literature in crystallized form, but only in one paper), and the reviewer claimed it was highly unlikely. We changed some of the more crazy parts for our transfer to ACS Catalysis and it got accepted there after the next round of review.
Dubey, A. Khaskin, E. 2016, ACS Catalysis, 3998-4002.
Although I had some positive feedback about the paper and there are a few enthusiastic citations (it hit the ACS Catalysis average I believe), it didn’t have the huge impact that I hoped for. I guess it is true that even if it’s a new reaction, that if it’s not practical for anyone, no one is going to be citing it. Still, whenever I draw the ester metathesis reaction on paper to someone for the first time, they look at it and say that it’s impossible, and then say that it’s really unusual. So, there is some satisfaction to be gained from that.
I had hoped to improve the metathesis process by having a more selective alcohol act as an acceptor of the acyl group, but there was no alternative that was cheap enough, and could drive the reaction to very high yields. You’re better off using ethanol and if the only way to get selective 99% alcohol formation from your ester is a few equivalents of a very expensive alcohol acceptor, then you’re actually better off with hydrogen gas probably. What might help is a catalyst that works even faster than the Gusev SNS catalyst. Then we could get away with loading more ethanol into the system and still have high yields of the alcohol after 1 day. Maybe even fatty acid esters would work. We do have a catalyst these days that works better and I can get much faster metathesis and higher yields (as of 2019) but it’s still not ideal. It’s not worth it to try to publish it just yet. I might return to this research project in the future and just recently I got some new catalysts to try out. For now however, it’s on hold.
The above was a bit long, but it’s still missing a few details probably, as I’m writing from memory a couple of years after the fact. If you are an interested chemist who has some suggestions on how to improve the reaction, and on the off chance that you managed to make it through the above text and our published paper, please get in touch.