

Research at OIST - Cyclopropanes from aliphatic alcohols
This entry has a throwback to the ester metathesis and transfer hydrogenation one. Remember when I said that this reaction reported by Milstein and Srimani will be talked about later and there will be a connection from the ester result? Well, this is that time.
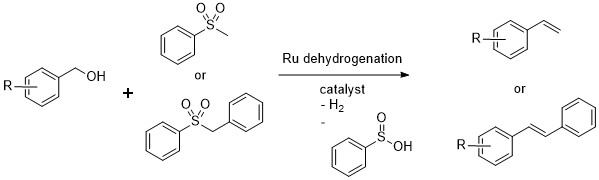
Dipankar’s reported reaction. Only the phenyl benzyl and phenyl methyl sulfones were reported
After the ester metathesis paper came out, I was thinking of what to do next with it and I kept trying new things to make it more selective, but because it’s a somewhat difficult problem (to which I will come back unless someone else does it (which looks unlikely since many people haven’t heard of it and it received an average number of citation for ACS Catalysis)) it just wasn’t in the cards if I’m only working on it five to ten hours a week, while also trying to do other interesting projects. One of the things that I am always doing on the side when I don’t have a catalysis project to focus on, is reaction screening. There was a paper by Hartwig where they screen a number of different substrates and catalysts in many wells at 100C and see what kind of interesting new products that they can get by GC/MS. Others have published similar things and there are of course plenty of papers that come about as a result of this type of screening where the authors don’t mention the background of the discovery.
Robbins, D.W.; Hartwig, J.F. A Simple, Multi-Dimensional Approach to High-Throughput Discovery of Catalytic Reactions. Science 2011, 333, 1423-1427.
The chemistry in these multi-welled reactors however, is in general relatively mild for Ru dehydrogenation catalysis conditions, involving reasonable temperatures close to room temperature, and often done under air. Of course, one of the big criticisms of Ru dehydrogenation catalysis is that it is unselective and will kill a lot of functional groups at 120 degrees, highly basic reflux in toluene for 24 hours under nitrogen. Unless your catalyst can make 10,000 TONs, then you’re making substrates for medicinal chemists and you need to have good functional group tolerance as is the case in many Pd and Ni couplings where the catalyst loading can be quite high. Even though there are now Ru catalysts that operate at low temperature of 60 degrees, they have only been tested for making the amide and the ester bond. The Wittig chemistry and Dipankar’s paper were early exceptions where carbon carbon bonds were formed. Although the functional group tolerance is not the best, it is surprisingly broad for such harsh conditions. Unfortunately, anything that the catalyst can latch onto (like primary and secondary amines, other non-tertiary OH groups you don’t want it to touch, alkenes that get isomerized, alkynes and cyano groups that get hydrogenated) is a problem and will lead to reduced yield. Playing around with the conditions means the alkynes and cyanos will survive and you’ll just get lower yields, but it’s a real problem if you can’t have other alcohols or amines in your molecule, as they make up a lot of drug space.
No surprise then, that blind screening with Ru dehydrogenation catalysts is not usually done. The other reason is that it’s not so simple to set up so many reaction wells under refluxing toluene conditions under nitrogen (especially if you never worked in the Hartwig group on this project I suppose). However, the fact that the Ru catalyst does latch onto those functional groups, is also an opportunity. And even if it doesn’t, as long as it just forms the aldehyde, that might be good enough for further reactivity. You have to have some sort of an idea when doing screening, so my idea for that particular screen, was to try the sulfones used by Dipankar together with esters. Back in the 2014 paper, they weren’t sure as to how the reaction actually proceeded, since if a sulfone adds to the intermediate aldehyde, it should form the Julia intermediate, which is pretty stable, and you need to introduce some sort of a radical reductant to induce sulfone elimination. The only thing that is available in the reaction mixture is hydrogen that comes from dehydrogenating the alcohol, so it was assumed that it was doing the role of a mercury amalgam in forming the olefin from the intermediate that was not isolated. Of course, if you start with an ester (just as in olefin metathesis), then you do not have any source of hydrogen and there will be no mystery reductant. So, I mixed together some unsymmetrical esters that I used in the metathesis work with benzyl phenyl sulfone (one of the two sulfones used in Dipankar’s paper), to see what would happen.
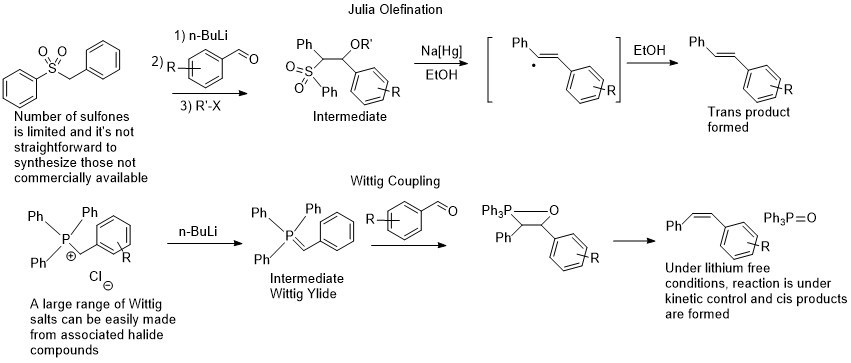
Julia and Wittig olefinations with the Julia intermediate that can be isolated shown
In retrospect, I was pretty lucky that I used the particular unsymmetrical esters that I did use. I’ll explain why in a minute. But basically, I got the olefin that was reported by Milstein and Dipankar in 2014 with benzyl acetate (i.e. I got stilbene), and I got three different products for hexyl acetate. Both products came out much later than the parent sulfone on the GC/MS (so the molecular mass had to be quite a few carbons more). The peak that was first was the minor product and the peaks that came out a few minutes later were close in retention time and had the same MW (so probably isomers). I isolated them on the column and took an NMR of each one.
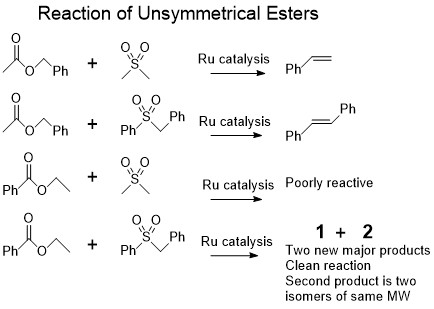
As often happens when you get something that you didn’t expect, the NMR doesn’t inspire an understanding of what the structure actually is. However, it does tell you some useful things. For example, the earlier product had two phenyl residues for each one ester alcohol residue, and I could tell that the heavier/later product had three phenyls to one alcohol. Since the sulfone used was benzyl phenyl sulfone with two phenyls, we could hypothesize that the first product was a one to one reaction of the sulfone and ester alkoxy residue and the later products were from a 2:1 reaction between the sulfone and the alkoxy unit, and involved some sulfone decomposition in the process, as one of the phenyls was missing. I could also tell that neither of the products had olefin CH peaks.
After getting this information and noting that there were some trace olefins in the case of ethyl benzoate, which probably resulted from ester metathesis before the sulfone could react with the alkoxy unit, I decided to use symmetrical esters such as hexyl hexanoate and ethyl acetate that would be unaffected by metathesis. The other change was that since the major products were 2:1, in order to get them in much larger yields, a 2:1 reactant ratio between the sulfone and the ester was used. These reactions did give much higher yields of the unknown products than the previous ones, and I was still able to get some of the first minor one that was the result of the 1:1 reaction in the case of hexyl hexanoate. After isolating them, it turned out that ethyl acetate gave crystals of the latter heavier 2:1 product and hexyl hexanoate gave crystals of the earlier 1:1 product. As the crystal structure tells you exactly what molecule you have, without having to guess from NMR data, this was great news. I was very surprised that the latter products turned out to be cyclopropanes, but even the 1:1 product was surprising since it was not an olefin and still had the sulfone attached. Both products were completely unexpected from following up on Dipankar’s paper. The later two peaks were diastereomers of the cyclopropane where the alcohol ended up either trans or cis to the remaining sulfone on the ring. One of them was always obtained in bigger amounts than the other and it depended on the alcohol residue. For hexanol from hexyl hexanoate, the ratio was about 3:1 for the diastereomers, but it was much bigger for ethanol from ethyl acetate.
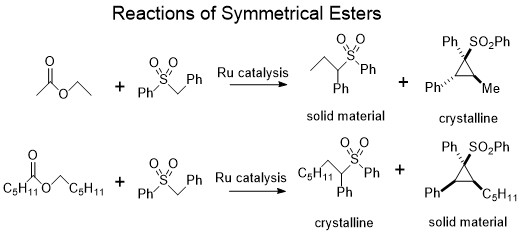
At this point I thought that there was now a very interesting reaction that could be optimized. I wanted to focus on the cyclopropanes because they seemed to be more valuable and interesting. The previous methods of cyclopropanes in the literature did not describe this type of a reaction. One of the most popular methods is generating a carbene by metal catalysis decomposition of a diazonium and adding it to a double bond. The closest method to our own seemed to be described by E.J. Corey where they used sulfur ylides to add to double bonds. However, I was not sure we had olefins involved in our process as there were no traces of them at all and we definitely did not have any sulfur ylides since sulfones are oxidation state VI and cannot form ylides. On the face of it, this was a completely new reaction that was not described in the literature. Another plus was that we took a simple and cheap sulfone and ester and made some weird, complicated three carbon cycle out of it that had a quaternary carbon center and three new stereocenters. And these types of cyclopropanes were also very different from the type that you can make by the most common metal catalyzed method as they require some sort of electron withdrawing group next to the olefin where the carbon would add. The type of cyclopropanes I had on hand were very electron rich.
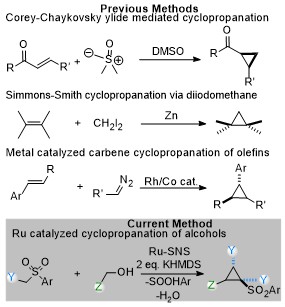
The downside of the reaction was that the yield was at the beginning not the ideal >90% required for publication in the journals that guarantee you tenure and that there were issues of obtaining the two diastereomers and not one selectively. I couldn’t control for which diastereomer was obtained… and also the reaction wasn’t enantioselective. Well, also the fact that it required a high temperature of 120C, and strong base was a minus as that would mean a lot of functional groups would have problems with it. Also, the Ru catalyst meant that functional groups that do react with it are incompatible. Still, all small prices to pay in light of describing a fundamentally new cyclopropanation reaction. At the time I thought that I would just have to show a little practicality in order to get a tenure positive journal to publish it.
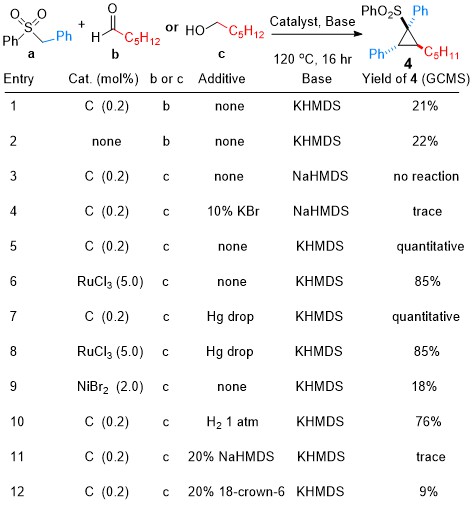
I did some optimization of the reaction just using GC/MS yields (this is very dangerous, but it can be an okay thing for optimization purposes). The first thing to do (besides using a 2:1 sulfone to ester ratio) was to get rid of the ester. Since only one part of it was reacting (the alcohol part) a lot of atoms were going to waste. I tried using only aldehyde or alcohol instead. The reaction worked and what’s interesting was that with the aldehyde it worked without any catalyst and I could still get ~20% yield. With the alcohol it didn’t work unless the catalyst was present, but if it was, the yields were much higher. The reason that the pure aldehyde worked so badly was that at high temperature and with strong base, it was disproportionating and probably some of those products were affecting the reaction as well. The reason this reaction was unknown to science before was because nobody would be crazy enough to throw aldehyde together with a substrate together a strong anion at 120 degrees and then try to look at what each one of those product peaks are (even if some of them are 20%). With the alcohol, the aldehyde was formed at a steady rate by Ru dehydrogenation and captured by the deprotonated sulfone before it could disproportionate together with a second aldehyde molecule. When this worked perfectly it was beautiful. The only thing I could see in the GC/MS was the internal standard peak and the two diasteromer product peaks. So technically the yield by GC/MS was quantitative. No hexanol based moieties either. I now realize that I should have made some allowance for byproducts that may not have been visible by GC/MS, but it was still very impressive. This also meant that the catalyst was not responsible for the actual formation of the cyclopropane and as long as we used something that can dehydrogenate the alcohol at high temperature, we should get cyclopropane. Indeed, it turned out that just some metal salts of metals that are known to form dehydrogenation catalysts will work, but it’s spotty. With RuCl3 you will see plenty of product, but also some garbage peaks, with NiBr2 it’s only about 18% product. Interestingly it turned out that the base was key. We needed to have a potassium base for this to work as Ni or Li ones gave zero percent yield. Maybe the potassium was helping to template the cyclization reaction?
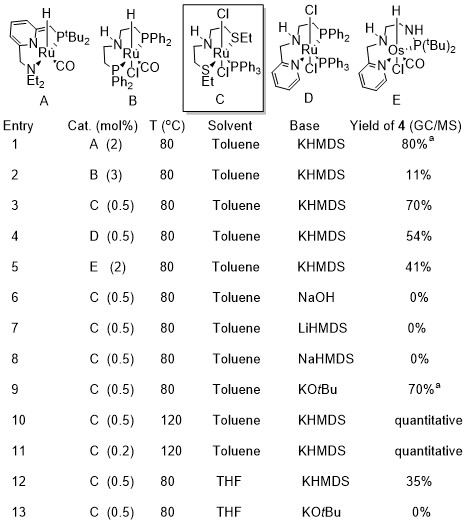
I then optimized the reaction for the catalyst. It turned out I was very lucky in choosing the Gusev catalyst based on the ester metathesis results. No other Ru dehydrogenation catalyst that I had lying around in the glove box was as selective for the cyclopropane. The other ones were less active (the osmium one), or more selective for the 1:1 linear product at lower temperatures. Even with the Gusev SNS catalyst I could see the linear product at lower temperature, but it was a lot less. The Milstein catalyst was actually very active and gave 80% cyclopropane already at 80C, but also more linear product than the Gusev catalyst, so I decided to go with the latter at 120C where it just gave pure cyclopropane. I think the reason the selectivity of the Gusev catalyst is that it works really well at dehydrogenating alcohol and it’s not stable at high temperatures. It quickly makes the aldehyde as required of it, and then decomposes before overstaying its welcome. My hypothesis at the time was that the linear product was formed by catalysts that were more stable that were doing things not asked of them.
At this time, an undergraduate intern, Tanner Jankins, joined the research group and signed up to work with me for five months. He had a lot of experience in synthetic labs before, and even did an internship at a pharma company, so I told him I had the perfect project that requires someone who can do a lot of column chromatography and NMR characterization. Over the next five months Tanner combined a lot of alcohols and sulfones at 120C in toluene with the Gusev catalyst at 1 mol% loading, and we learned where our reaction worked well and where it didn’t. I told Tanner to try and isolate only one of the diastereomers by chromatography if he could manage it (the polarity differences were not that high for many of them) and we sort of managed it by throwing away some mixed fractions. So, the hexanol based cyclopropane went from 3:1 in the crude mixture to 3.6:1 in the pure form for example, but the ethanol based cyclopropane went from 12:1 to basically no trace of the minor diastereomer (99:1) after the column. Some of the results were really frustrating. Such as compound 12 where we got quantitative yield by GC/MS and could see a huge amount of the product by crude NMR, but just got so little from the column. I repeated that reaction myself and had the same failure. We got only ~30% isolated product. I now realize that we were not as good at hand columns as we thought, and we should have used a gradient with some ammonia in it. Now that we have an automated flash and I learned how to use basic gradients, amines are no longer as much of a problem.
In general, the yields are a bit low and usually below 50%. However, making these complicated cyclopropanes by any other way takes a lot of steps with lower overall yields and our starting materials (sulfone for 1 dollar a gram and alcohol for 1 dollar for 20 grams or more) can’t be beat. Even if a yield for this reaction is 10%, our method is still more cost effective. The problem is, since no one ever made these sulfones before due to how difficult they were to make previously, does anyone care about the cost if they don’t know what they need them for? Still, this is a new reaction so it should not really matter. Or so I thought.
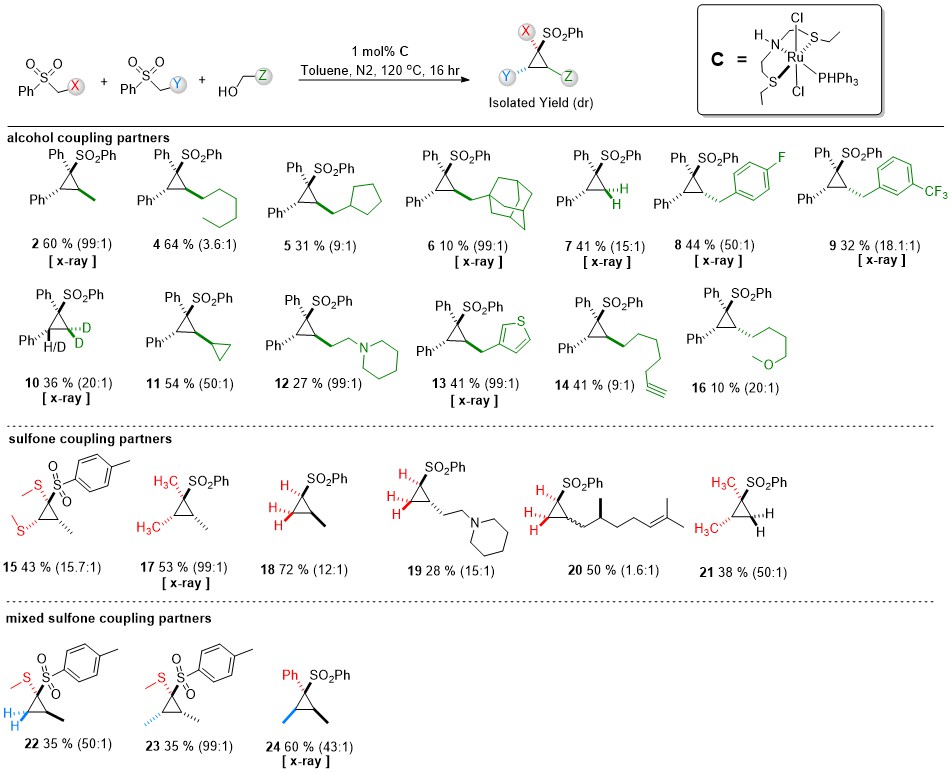
Tanner found that sterics mattered a lot for the outcome of the reaction. If the alcohol was sterically demanding, like a secondary one or had a really big group on the beta carbon, then the reaction simply did not take place to even a 10% yield extent. However, having substitution on the gamma carbon was fine. For a lot of the substrates, having heteroatom substitution on the beta carbon led to some sort of rearrangement decomposition. For a long time, we were surprised by the reaction of butynols where there was a triple bond on the beta or gamma carbon. It gave the same product as just formaldehyde without catalyst (entry 7) and eventually we figured out that after the aldehyde was formed it did a retro-Reformatsky rearrangement to form formaldehyde. It was possible to incorporate a triple bond, but only if it’s far away from the alcohol. Hydrogenation of olefins and triple bonds at this high temperature by the Ru catalyst is a problem, but if the substrate is the alkoxy part of an ester, then hydrogenation does not occur.
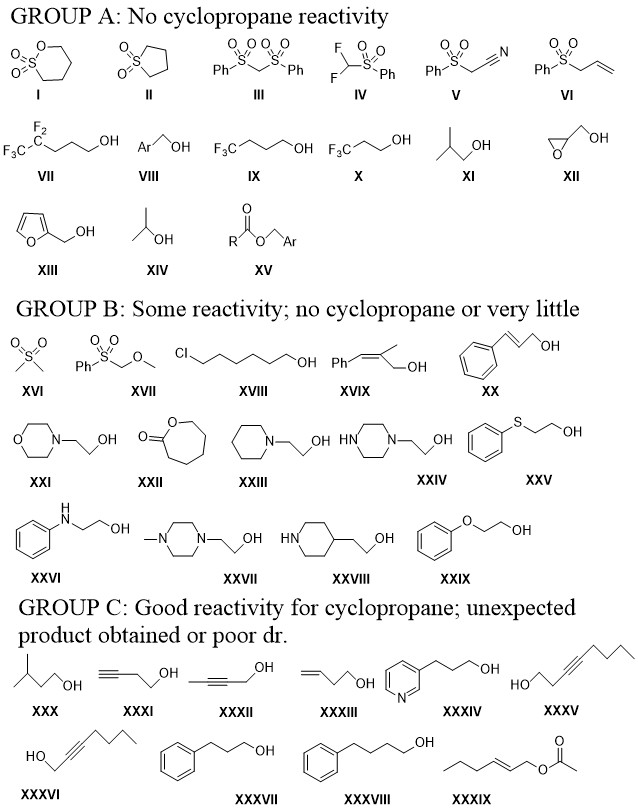
A lot of the substrates could crystallize, and this was really helpful in determining the relative position of all the groups around the cyclopropane ring, as only determining it from NMR NOESY data is a bit risky.
For the mechanism, I did a few reactions which tried to establish the intermediacy of olefins in the reaction, but they all seemed to suggest that olefins are not involved. Reviewers did mention that maybe the vinyl sulfone polymerizes too fast, but this is unlikely as I would still expect to see some trace of product. Also, addition to the olefin to give cyclopropane requires a carbene normally, and it’s hard to see how an anion of another sulfone can form a carbene.
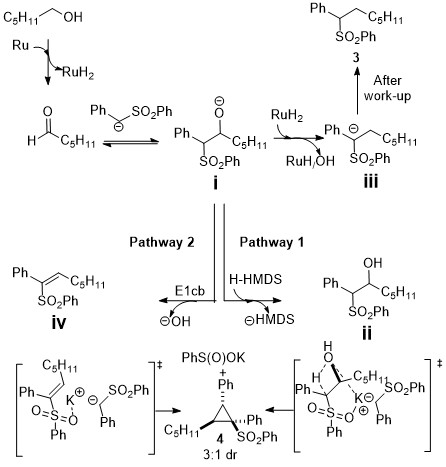
As you can guess if you read some of my previous entries, the reviewers were not happy at all with the mechanism and kept insisting an olefin was involved. I drew it in as potential pathway 2, but then argued that it probably did not occur this way. The thing that got the most comments and righteous anger from the reviewers during review was the mechanism. The second thing that bothered at least one reviewer all the time was the low yields. I argued that it didn’t matter because: the starting materials are cheap; we’re saving 4-7 steps; this is a completely new reaction; we form very complex things from something very simple; etc… but it didn’t help. Of course, it’s a new reaction that looks unusual so at least it went out for review in all the flagship journals, but it still got rejected. I even tried the trick of putting the GC/MS yields in and the actual isolated yields in brackets right beside them, so it looked more impressive, but it turns out the reviewers actually read the article in detail and didn’t really care/fall for it. At least it means that it was interesting to them I suppose. The editor of JACS told me to submit it to Organic Letters but out of being irrationally upset at the rejection from there, I decided not to listen to him and submit it to Organometallics, which is a journal that I actually read, unlike OL (which does have a higher impact factor though). It got accepted to Organometallics and the editor who was taking care of it told me they really liked it when I met them at a conference. I realize now that the audience for the paper is better matched to OL though.
“Three-Component [1 + 1 + 1] Cyclopropanation with Ruthenium(II)” Tanner C. Jankins; Robert R. Fayzullin; Eugene Khaskin, Organometallics, 2018, 2609-2617.
Based on this reaction, we were actually able to design a few more new ones. With the lessons learned from this article I was able to find a few more interesting transformations. The most obvious one is the linear sulfones of course. Are we able to fine tune the conditions to isolate the first 1:1 reaction product as the major one without getting any cyclopropane? Well, we are. That was the next most obvious thing to try. But there are a couple more things that are not so obvious… I’ll write an entry for each of those reactions once the relevant paper is published, including the 1:1 linear sulfone product.