

FY2012 Annual Report
G0 Cell Unit
Professor Mitsuhiro Yanagida

Abstract
Regulation of cellular proliferation and quiescence (G0 phase) in response to environmental changes, such as the availability of nutrients, is a central issue in biology and medicine. Our research goal is to elucidate the molecular mechanisms that regulate pivotal events in cellular proliferation and quiescence, such as chromosome segregation, coupling of cell growth and division, and cellular adaptation to nutrient paucity. To study the molecular mechanisms in detail, we have adopted the fission yeast Schizosaccharomyces pombe, a simple unicellular eukaryote, as a model organism, and use post-genomic methods in combination with genetic approaches. This year, we further evaluated the G0 phase induced by nitrogen starvation and the adaptation of S. pombe to glucose-limited conditions. We also improved the methodology for metabolomic studies and developed a comprehensive software algorithm for chemical formula prediction from mass spectrometry data. In addition, we investigated various regulatory mechanisms of proliferation. Some relations between metabolic synthesis and cell cycle progression were revealed by combined metabolomic and genetic analyses. We reported a study of the coenzyme A biosynthesis pathway. We also investigated the health benefits of Okinawan produce and the backgrounds of Okinawan longevity as one of the R&D cluster projects in Okinawa. We published one review article and three original research articles in FY2012.
1. Staff
- Dr. Takeshi Hayashi, Group leader
- Dr. Kojiro Takeda, Group leader
- Dr. Kenichi Sajiki, Researcher
- Dr. Norihiko Nakazawa, Researcher
- Dr. Takayuki Teruya, Researcher
- Dr. Xiaodong He, Researcher
- Dr. Emily Tsang, Researcher (from March)
- Ms. Ayaka Mori, Technical staff
- Mr. Tomas Pluskal, Technical staff
- Ms. Yuria Tahara, Technical staff
- Mr. Masahiro Ebe, Technical staff
- Ms. Orie Arakawa, Technical staff
- Ms. Caroline Starzynski, Technical staff (from June, until September)
- Ms. Chikako Sugiyama, Research administrator
- Mr. Xingya Xu, Special Research Student
- Ms. Flora Smit, Research intern (from October)
2. Collaborations
- Theme: Metabolite structure determination using NMR
- Type of collaboration: Joint research
- Researchers:
- Professor, Masaru Ueno, Department of Molecular Biotechnology, Graduate school of Advanced Science of Matter, Hiroshima University
- Theme: Identification of fission yeast mutants, which show growth defect under low glucose condition
- Type of collaboration: Joint research
- Researchers:
- Professor, Shigeaki Saitoh, Institute of Life Science, Kurume University
- Theme: Identification of the molecular mechanism, required for the maintenance of cell viability in low glucose condition
- Type of collaboration: Joint research
- Researchers:
- Professor, Kunihiro Ohta, Department of Life Science, Graduate School of Arts and Science, University of Tokyo
- Theme: Metabolomic analysis of human blood
- Type of collaboration: Joint research
- Researchers:
- Dr. Hiroshi Kondoh, Department of Geriatric Medicine, Graduate School of Medicine, Kyoto University
- Dr. Takumi Mikawa, Department of Geriatric Medicine, Graduate School of Medicine, Kyoto University
- Theme: Metabolic analysis of active ingredients of health foods
- Type of collaboration: Joint research
- Researchers:
- Dr. Hiroshi Kondoh, Department of Geriatric Medicine, Graduate School of Medicine, Kyoto University
- Theme: Metabolic analysis of active ingredients of health foods
- Type of collaboration: Joint research
- Researchers:
- Professor Hiroaki Masuzaki, Division of Endocrinology, Diabetes and Metabolism, Hematology, Rheumatology (Second Department of Internal Medicine), Graduate School of Medicine, University of the Ryukyus
- Theme: Dolphin blood metabolome analysis
- Type of collaboration: Joint research
- Researchers:
- Dr. Tadashi Maruyama, Marine Biodiversity Research Program, Japan Marine Science and Technology Center (JAMSTC)
- Mr. Yoshiaki Kamei, Okinawa Churaumi Aquarium, Okinawa Churashima Foundation
3. Activities and Findings
3.1 Mutant screening of genes required for the G0/quiescent phase
We have been searching for genes that are essential for transition between the growth phase and the G0 phase. In previous viability screenings under nitrogen starvation conditions, we identified 86 and 102 genes that are required for G0 phase survival from a temperature-sensitive (ts) mutant library and a deletion mutant library, respectively. These genes were identified using whole genome sequencing technology together with backcrossing techniques. Among these genes, we identified cdc2. The cdc2 ts mutant was named cdc2-974 and these cells failed to form small and round G0-like cells after nitrogen starvation, even at 26ºC. The mutation site of this strain located at I35N in cdc2, and we confirmed that this is the responsible mutation site by integrating the same mutation in wild-type (WT) cells. The integrated cells exhibited decreased viability and an elongated cell shape after nitrogen starvation, confirming that this mutation is responsible. Further analysis identified that the cdc2-974 mutant failed the second division after nitrogen starvation and remained in the elongated shape at 26ºC, which is a trait observed only in this cdc2 mutant allele (Figure 1).
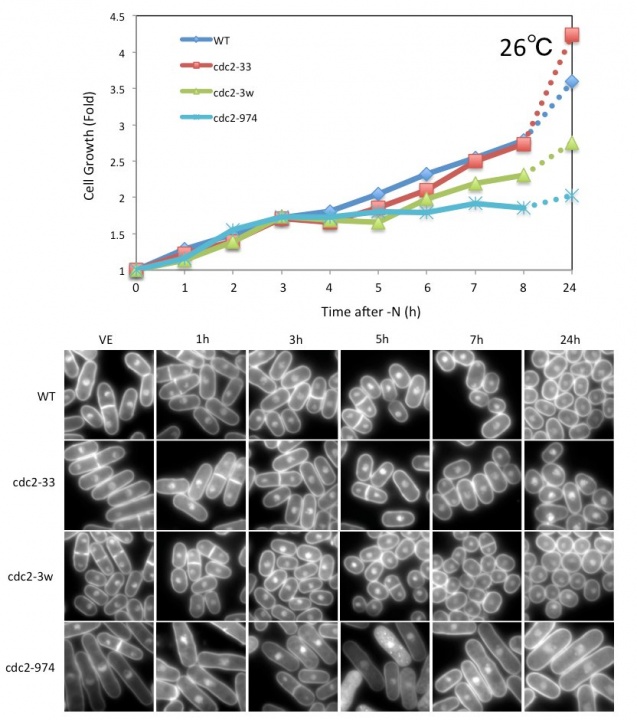
Figure 1. Growth curve and DAPI images of WT and cdc2 mutant strains after nitrogen starvation. The cdc2-974 strain divided once and exhibited an elongated shape after nitrogen starvation at 26˚C.
Several strains identified from the screening showed defects like the cdc2 mutants at G0 entry. Therefore, we performed a liquid chromatography-mass spectrometry (LC-MS) experiment immediately after nitrogen starvation to analyze the cellular profiles at the metabolome level. We obtained samples before and 15, 30, and 60 min after nitrogen starvation, and repeated the experiment three times. From these experiments, approximately 3600 peaks were detected and 291 metabolites were identified. Among these metabolites, we identified 40 metabolites whose amounts were significantly changed immediately after nitrogen starvation (Figure 2). These metabolites could be good biomarkers for the screened mutants. We also performed a metabolome analysis of longer time points after nitrogen starvation and are currently analyzing the data.
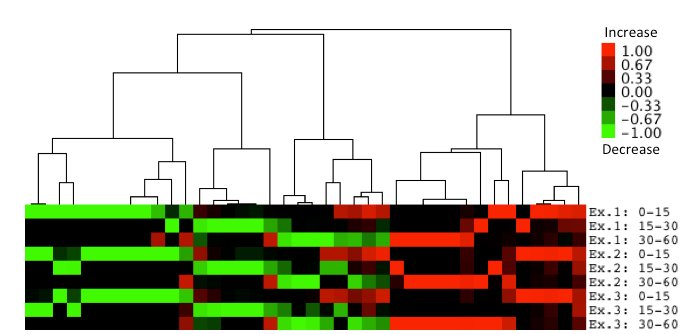
Figure 2. Identification of 40 metabolites whose amounts were changed immediately after nitrogen starvation. Metabolites whose absolute values of the amount of change in log2 are equal to or greater than 2.0 in at least one of three intervals are clustered and shown in tree view images. The log2 values are normalized and increased values are shown in red and decreased values are shown in green. 40 metabolites exhibiting the same and similar behaviors in all three experiments were extracted and clustered hierarchically using a complete linkage method.
3.2 Development of a comprehensive software algorithm for chemical formula prediction from mass spectrometry data
Mass spectrometry is commonly applied to qualitatively and quantitatively profile small molecules such as peptides, metabolites, or lipids. Modern mass spectrometers provide accurate measurements of mass-to-charge ratios of ions, with errors as low as 1 ppm. Even such high mass accuracy, however, is not sufficient to determine the unique chemical formula of each ion, and additional algorithms are necessary. We developed a universal software tool to predict chemical formulas from high-resolution mass spectrometry data (Figure 3A). The tool was implemented within the MZmine 2 mass spectrometry data processing framework (Pluskal et al., 2010). The algorithm is based on the use of a combination of heuristic techniques, including MS/MS fragmentation analysis and isotope pattern matching (Figure 3B). The most selective filter to obtain the best candidate chemical formulas was based on isotope pattern comparison, the algorithm for which was newly developed (Figure 3C) and outperformed a previously published method in 64% of the tested Orbitrap spectra. The performance of the tool was evaluated using a real metabolomic dataset obtained with the Orbitrap MS detector. On average, the heuristic algorithms reduced the search space for potential formula candidates 40-fold (Figure 3D). The true chemical formula was correctly determined as the highest-ranking candidate for 79% of the tested compounds. This software algorithm was published in Analytical Chemistry in May 2012.
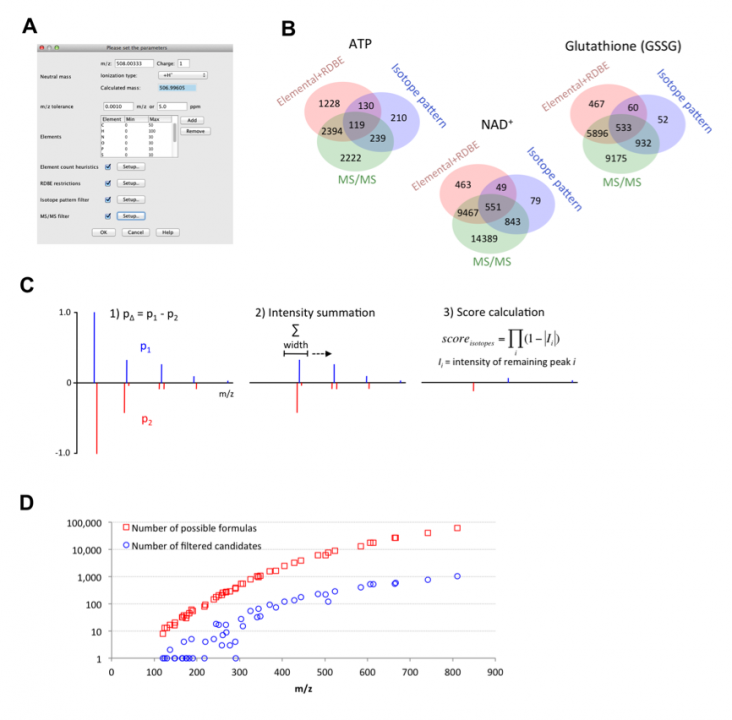
Figure 3. A. Parameter setup dialog of the formula prediction tool. B. Venn diagrams showing the efficiency of the different types of heuristics for the prediction of formulas for ATP, glutathione (GSSG), and NAD+ ions. The red circle shows the number of generated formulas that passed through the elemental ratios and RDBE filters. The blue circle shows the number of formulas that passed through the isotope pattern-scoring filter (minimum score 80%). The green circle shows the number of formulas that passed through the MS/MS filter (minimum score 100%). C. The algorithm used to calculate the score of the isotope pattern similarity. D. Scatter plot of 48 analyzed compounds with m/z value on the X-axis and number of possible formulas (red square) or number of filtered candidates (blue circle) on the Y-axis.
3.3 Study of the coenzyme A biosynthesis pathway in Schizosaccharomyces pombe
Biosynthesis of coenzyme A (CoA) requires a five-step process utilizing pantothenate and cysteine in the fission yeast S. pombe (Figure 4A). CoA contains a thiol (SH) group that reacts with carboxylic acid to form thioesters, giving rise to acyl-activated CoAs such as acetyl-CoA. Acetyl-CoA is essential for energy metabolism and protein acetylation, and in higher eukaryotes it is essential for the production of neurotransmitters. We isolated a novel S. pombe ts strain, ppc1-537, mutated in the catalytic region of phosphopantothenoylcysteine synthetase (PPCS), which is essential for CoA synthesis (Figure 4 B and C). The mutant becomes auxotrophic to pantothenate at a permissive temperature (Figure 4D), and has greatly decreased levels of CoA, acetyl-CoA (Figure 4E), and histone acetylation. Moreover, ppc1-537 mutant cells failed to restore proliferation from quiescence (Figure 4F). Ppc1 is thus the product of a superhousekeeping gene. The ppc1-537 mutant showed combined synthetic lethal defects with five of six histone deacetylase deletion mutants, whereas sir2 deletion exceptionally rescued the ppc1-537 phenotype. In synchronous cultures, ppc1-537 cells can proceed to the S phase, but loses viability during mitosis, failing in sister centromere/kinetochore segregation and nuclear division. Additionally, double strand break repair is defective in the ppc1-537 mutant (Figure 4G), producing fragile broken DNA, probably due to diminished histone acetylation. The CoA-supported metabolism thus controls the state of chromosome DNA. This study was published in Open Biology in September 2012.
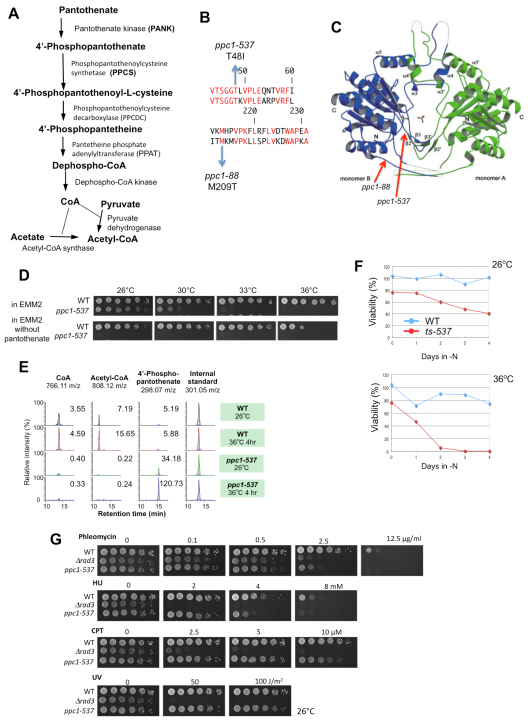
Figure 4. A. The biosynthetic pathway of CoA from pantothenate. B. The substitution mutation sites (T48I and M209T) determined for ts-537 and ts-88 strains, respectively, reside within the coding region of PPCS/Ppc1 (top, S. pombe; bottom, human sequence). Identical residues are in red. C. Location of mutation sites in the three-dimensional structure of human PPCS. D. Pantothenate is needed for ppc1-537 but not for the wild-type (WT). Spot tests were done for the WT and ppc1-537 in the presence or absence of pantothenate. E. WT and ppc1-537 grown at 26°C were transferred to 36°C for 4 h in the EMM2 medium. Metabolites were extracted from WT and mutant cells and analyzed. Levels of CoA, acetyl-CoA, and 4’-phoshpopantothenate are shown. F. WT and ts-537 cells were brought into the G0 quiescent state at 26°C under nitrogen source deficiency for 24 h. Resulting G0 quiescent cultures were shifted to 36°C for 4 days. As the control, portions of the cultures were kept at 26°C. G. WT, ppc1-537, and ∆rad3 mutant cells were grown at 26°C and spotted on rich YPD medium containing the DNA-damaging agents phleomycin, hydroxyurea (HU), or camptothecin (CPT), or irradiated by ultraviolet rays (UV), and cultured at 26°C for 4 days.
3.4 Impaired phosphatidylcholine synthesis and aberrant nuclear division in mutants of SAM and SAH cycle enzymes
In a previous study, we found that S-adenosylmethionine (SAM or AdoMet) strongly increased (~20-fold) at glucose concentrations below 2.2 mM (0.04%) (Pluskal et al., 2011). SAM is an important compound related to many cellular functions because SAM serves as the methyl donor in most methyl transfer reactions, including methylation of proteins, nucleic acids and lipids. SAM is converted to S-adenosylhomocysteine (SAH) during methyl transfer reactions and then SAH is hydrolyzed into homocysteine and adenosine (Figure 5A). Because SAH is a potent product inhibitor of methyltransferase reactions, the SAM/SAH ratio is considered an indicator of the cellular methylation capacity. Therefore, regulation of the synthesis of SAM and hydrolysis of SAH could affect cellular methyl transfer reactions. Because cellular defects in the genetic disruption of SAM synthesis or SAH hydrolysis are not well understood, we isolated ts mutants of SAM synthetase (Sam1) and SAH hydrolase (Sah1) in S. pombe by a PCR-based random mutagenesis screen. sam1 and sah1 mutants showed the lsd (large and small daughter nuclei) phenotype in mitosis at a restrictive temperature: DAPI-stained chromatin regions in the daughter nuclei differed in size (Figure 5B). Sister chromatids, however, appeared to be separated in cells showing the lsd phenotype. This phenotype resembles that of the mutant of fatty acid synthase Fas2. Metabolomic analysis revealed that the amount of some metabolites related to phospholipid metabolism dramatically decreased or increased in sam1-1 mutant cells. De novo phosphatidylcholine (PC) synthesis is reported to require three SAM-dependent methylation steps. Taken together, PC synthesis seems to be impaired in sam1 and sah1 mutants. Furthermore, we found that the lsd phonotype of sam1 and sah1 mutants was suppressed by choline, a precursor of PC in the methylation-independent pathway, suggesting that the lsd phenotype is caused by a PC synthesis defect.
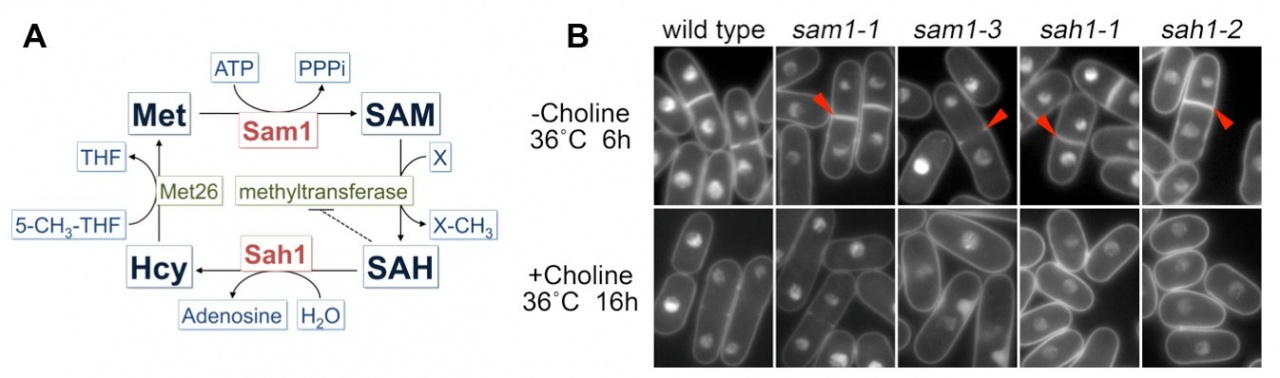
Figure 5. A. Schematic illustration of the methionine cycle in fission yeast. B. Cellular phenotype of sam1 and sah1 mutants stained with DAPI. Arrowheads indicate binucleate cells displaying unequal nuclear division.
3.5 Condensin accumulation at the transcriptional active genes
Condensin plays essential roles in chromosome dynamics and diverse DNA metabolism. Previously, we demonstrated that condensin removes replication protein A (RPA) and RNA molecules from single-stranded DNA through its reannealing activity in vitro. How the molecular activities of condensin contribute to chromosome segregation, however, is largely unknown. Here, we report that S. pombe condensin accumulates at highly transcriptional active sites in mitosis (Figure 6). This accumulation is more prominent in chromatin immunoprecipitation-sequencing (ChIP-seq) of condensin (Cut14/SMC2), than in that of cohesin (Rad21) and topoisomerase II (Top2) proteins. Consistent with the continuous requirement for condensin throughout mitosis, condensin de novo associates with the heat-shock responsible genes from metaphase to anaphase and gradually accumulates around the 3’-UTR region. These and other results suggest that condensin in mitosis associates with the highly transcribed regions and may resolve the RNA- and/or protein-bound obstructive sister chromatid DNAs into the smoothly separable chromatid DNAs for faithful chromosome segregation during mitosis.
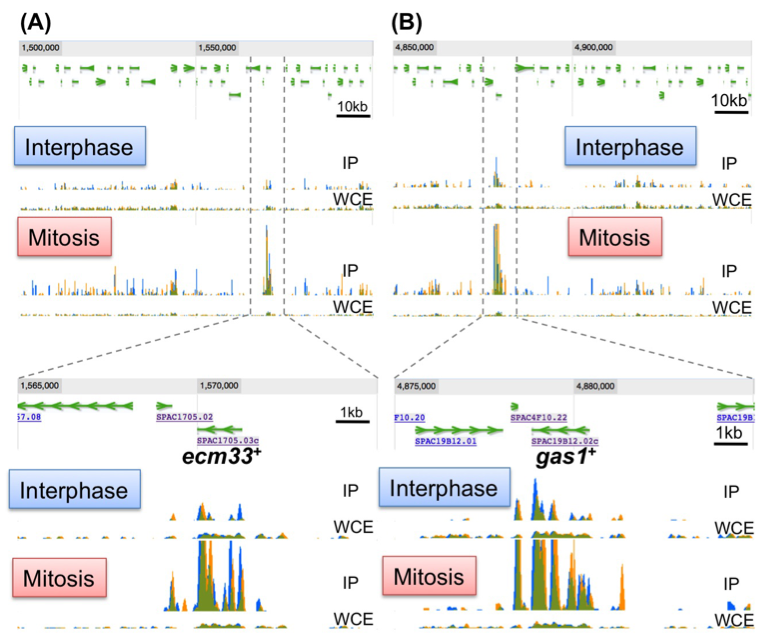
Figure 6. Condensin accumulates at the highly transcribed genes in mitosis. Identification of condensin association sites by a ChIP-seq experiment. The ecm33+ (A) and gas1+ (B) genes show periodic expression in mitosis and encode the cell wall component. IP: Immunoprecipitated sample, WCE: whole cell extract sample.
3.6 Genetic control of glucose starvation response in fission yeast
Glucose is a crucial source of cellular carbon and energy in most organisms, and regulation of its proper uptake and utilization is a key challenge for cell growth control. Fission yeast S. pombe cells can grow and divide even in 0.02% low glucose conditions, and proliferate at basically the same constant rate under a broad concentration range of glucose (0.08%-2%). To understand how S. pombe cells adapt their cellular growth and division in low glucose concentrations, we identified 170 and 49 low glucose survival (lgs) genes that were essential for proliferation in low glucose conditions (0.02%-0.08%) from a deletion mutant library and a ts mutant library, respectively. We found that among eight hexose transporters in S. pombe, only Ght5 is essential for proliferation in low glucose. We also elucidated that TORC2-PDK1-AKT signaling is required for proper cell surface localization of Ght5 in low glucose, whereas AMPK, CaMKK-like Ssp1, and PP2A-like phosphatase inhibitor Sds23 are required for the elevated expression of Ght5 through the nuclear exclusion of zf repressor Scr1 under low glucose conditions (Fig. 7). Further functional analyses on isolated mutants are in progress (in collaboration with Kurume University).
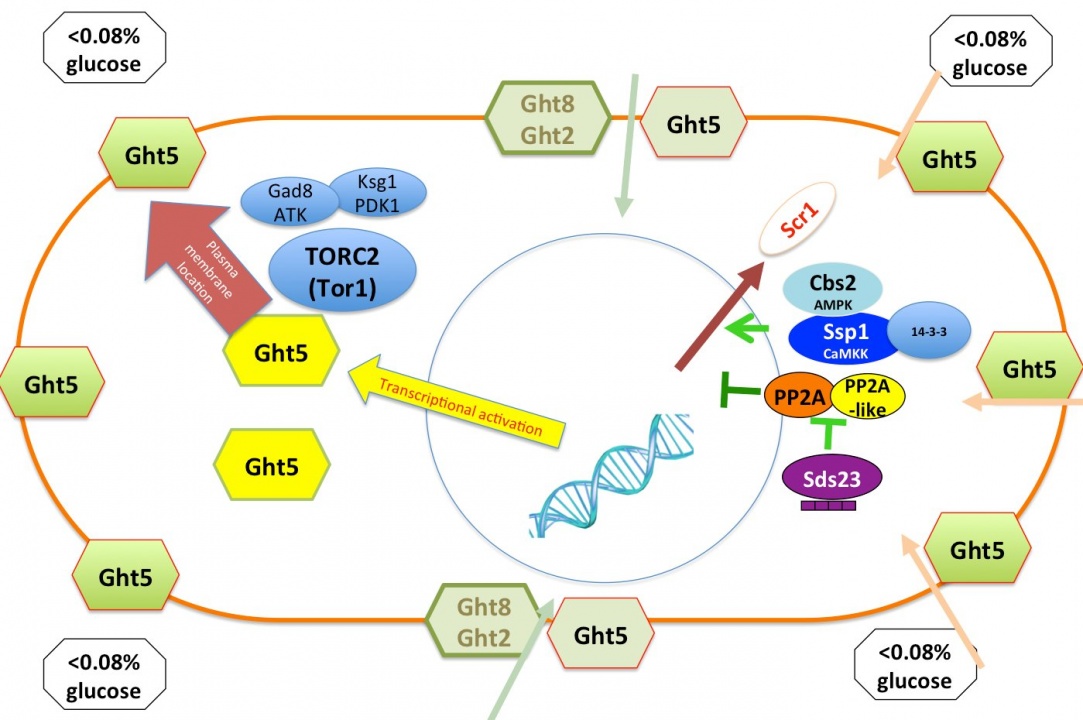
Figure 7. Two events ensure efficient glucose uptake in low glucose conditions. Transcription of the major glucose transporter (Ght5) is up-regulated by AMPK, CaMKK-like Ssp1, and PP2A-like phosphatase inhibitor Sds23 through the nuclear exclusion of zf repressor Scr1. On the other hand, the accumulation of newly synthesized Ght5 and other transporters along the whole cell surface requires TORC2-PDK1-AKT signaling.
3.7 R&D cluster projects in Okinawa
Our research proposal, ‘Metabolomic study for the technological development of health-longevity improvement through new approaches to the transdermal absorption of active ingredients’, was launched as an R&D cluster project in Okinawa in 2011. Two laboratories at the Ryukyu and Kyoto Universities, and two private companies, SOMNOQUEST and Sentan Iryou Kaihatu, joined together to form a research team that will use an analytic technology called “metabolome analysis” to search for Okinawan products that contain substances to treat disease, and to conduct research on the transdermal absorption process of such substances. We have begun to examine the metabolism of active ingredients and their effect on human blood metabolism after absorption of Okinawan medicinal plants (in collaboration with SOMNOQUEST and Kyoto University). We have also begun to analyze the blood metabolome of diabetic and non-diabetic people (in collaboration with University of the Ryukyus).
Furthermore, we will use proteomics to complement data derived from metabolomics to assist in providing a systems approach to study human health and disease. In a previous study, we identified a lot of metabolomic compounds in human blood. Now we will use mass spectrometry based proteomics to characterize the mouse red blood cell (RBC) protein profile to find correlation enzyme blood metabolites. RBC proteomic data have been used to reconstruct the most expansive description of RBC metabolism to date, following extensive manual curation, literature review, and functional testing. The reconstruction contains 235 enzymes representing functions from glycolysis to cofactor and amino acid metabolism. There are 10 enzymes catalyzing glycolysis (glucose ➡ pyruvate, Figure 8). The first five steps are considered to comprise the preparatory (or investment) phase, as they consume energy to convert the glucose into two three-carbon sugar phosphates. The next five steps of glycolysis comprise the pay-off phase, characterized by a net gain of the energy-rich molecules ATP and NADH. Thus, our analysis of the RBC proteome has revealed an unexpected level of complexity in the functional capabilities of RBC metabolism.
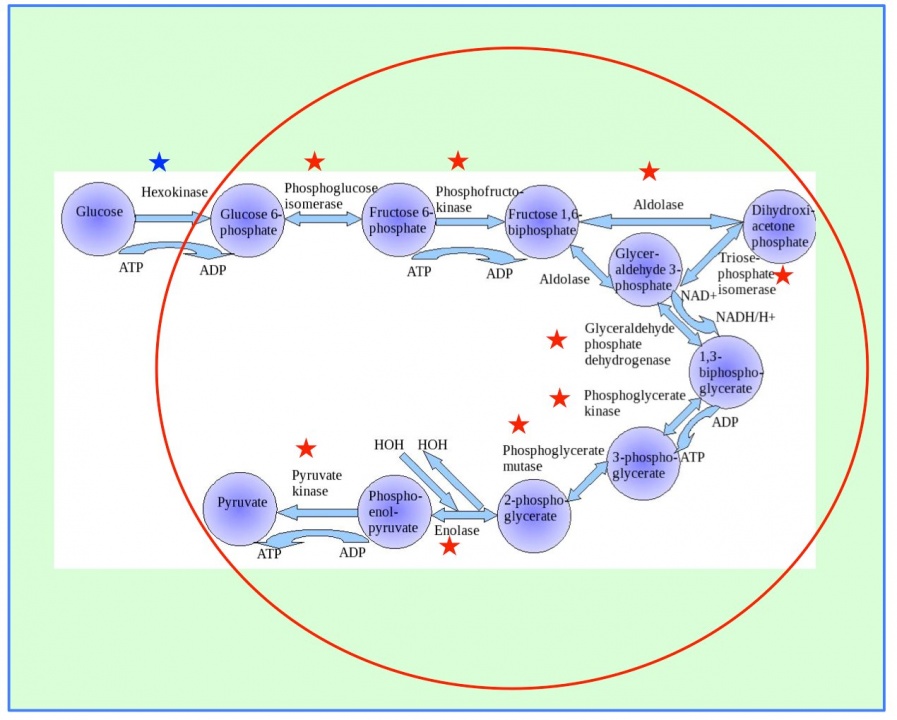
Figure 8. Ten enzymes catalyze glycolysis (glucose ➡ pyruvate). The first step (blue star) is catalyzed in plasma, and the other nine steps (red stars) are in the RBCs.
4. Publications
4.1 Journals
- Klionsky, D. J. and 1269 authors including Yanagida, M. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445-544 (2012).
- Nakamura, T., Pluskal, T., Nakaseko, Y. & Yanagida, M. Impaired coenzyme A synthesis in fission yeast causes defective mitosis, quiescence-exit failure, histone hypoacetylation and fragile DNA. Open biology 2, 120117, doi:10.1098/rsob.120117 (2012).
- Pluskal, T., Uehara, T. & Yanagida, M. Highly accurate chemical formula prediction tool utilizing high-resolution mass spectra, MS/MS fragmentation, heuristic rules, and isotope pattern matching. Analytical chemistry 84, 4396-4403, doi:10.1021/ac3000418 (2012).
- Tay, Z., Eng, R. J., Sajiki, K., Lim, K. K., Tang, M. Y., Yanagida, M. & Chen, E. S. Cellular robustness conferred by genetic crosstalk underlies resistance against chemotherapeutic drug doxorubicin in fission yeast. PLoS One 8, e55041, doi:10.1371/journal.pone.0055041 (2013).
4.2 Books and other one-time publications
Nothing to report
4.3 Oral and Poster Presentations
- Yanagida, M. The Role of Condensin in centromere/kinetochore during mitosis, in EMBO Workshop 'Structure, Function, and Regulation of Centromeres and Kinetochores', Barcelona, Spain (2012 ).
- Yanagida, M. DNA damage repair metabolism in dividing and non-dividing cells in The 3rd International Society of Radiation Neurobiology Conference, Bankoku Shinryokan, Okinawa (2013).
- Nakazawa, N. Condensin Accumulation at the Transcriptional Active Genes during Mitosis, in 6th Retreat Meeting of the IMSUT & RCAST Global COE OIST, Okinawa (2012).
- Pluskal, T. MZmine 2 – a modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data, in The 7th Metabolome Symposium, Keio University, Tokyo (2012).
- Yanagida, M. Study of Human Longevity using Fission Yeast as a Model and Red Blood Cell as a Marker, in New Frontiers of Metabolism Research in Biomedical Sciences The University of Tokyo, Tokyo (2012).
- Yanagida, M. Control of Cellular Quiescence, in Cell Inovation-OIST Joint Meeting, OIST, Okinawa (2013).
- Nakazawa, N. Casein Kinase 2-dependent Constitutive Phosphorylation of Fission Yeast DNA Topoisomerase II at C-terminal Charged Region, in 35th Annual Meeting of Japan Molecular Biology Society, Fukuoka International Conference Hall, Fukuoka (2012).
- Yanagida, M. How to conduct creative research?, in 52nd symposium for young biochemist researcher Hotel Tatsuki, Aichi (2012).
- Yanagida, M. Metabolomic study for the technological development of health-longevity improvement through new approaches to the transdermal absorption of the active ingredients, in Symposium "Establishing a research hub toward the development of an intellectual cluster", Pachific Hotel Okinawa, Okinawa (2012).
- Yanagida, M. Is the yeast metabolome useful for the research of human longevity and ageing?, in The 20th Yeast Joint Symposium Kyoto University, Kyoto (2012).
- Pluskal, T. Methods for metabolomics data processing, storage and analysis, in Symposium "Establishing a research hub toward the development of an intellectual cluster", Pacific Hotel Okinawa, Okinawa (2012).
- Pluskal, T. Highly accurate chemical formula prediction tool utilizing high-resolution mass spectra, MS/MS fragmentation, heuristic rules, and isotope pattern matching, in The 19th International Mass Spectrometry Conference, Kyoto (2012).
- Sajiki, K. Comprehensive screening of S.pombe deletion mutant libraries for essential genes under cellular quiescence, in Yeast Geneticis & Molecular Biology Princeton University, New Jersy, USA (2012).
- Ebe, M. Goat and dolphin blood Metabolome analysis, in Symposium "Establishing a research hub toward the development of an intellectual cluster", Pacific Hotel Okinawa, Okinawa (2012).
- Hayashi, T. Aberrant mitosis in fission yeast mutants defective in methionine-metabolizing enzymes, in The 30th Chromosome Workshop, The 11th Nuclear Dynamics Meeting, Awaji Yumebutai International Conference Center, Hyogo (2012).
- Hayashi, T. Characterization of methionine-metabolizing enzymes in fission yeast, in The 1st Conference of Molecular Cell Pathology for Lifestily Related Disease Royal Park Hotel, Tokyo (2013).
- He, X. Metabolomics complements data derived from proteomics, in Symposium "Establishing a research hub toward the development of an intellectual cluster", Pacific Hotel Okinawa, Okinawa (2012).
- Nakazawa, N. Fission yeast condensin de novo accumulation at the transcriptional active genes even during metaphase, in The 30th Chromosome Workshop, The 11th Nuclear Dynamics Meeting, Awaji Yumebutai International Conference Center, Hyogo (2012).
- Nakazawa, N. Casein Kinase 2-dependent Constitutive Phosphorylation of Fission Yeast DNA Topoisomerase II at C-terminal Charged Region, in 35th Annual Meeting of Japan Molecular Biology Society, Fukuoka International Conference Hall, Fukuoka (2012).
- Teruya, T. The analysis of changes in human blood metabolome caused by intaking Artemisia indica Willd, in Symposium "Establishing a research hub toward the development of an intellectual cluster", Pacific Hotel Okinawa, Okinawa (2012).
- Pluskal, T. Comprehensive Metabolomic Analysis of the Fission Yeast and Its Application to Glucose Starvation, Hiroshima University, Hiroshima (2012).
- Pluskal, T. Comprehensive Metabolomic Analysis of the Fission Yeast and Its Application to Glucose Starvation, Kurume University, Fukuoka (2012).
- Yanagida, M. Approaches for Human Longevity and Aging using Fission Yeast, Institute of Molecular Biology Taipei, Taiwan (2012).
- Yanagida, M. Genetics and Epigenetics of Glucose Starvation in Fission Yeast, Cambridge, UK (2012).
- Yanagida, M. What we will understand from the metabolomic analysis?, Kazan Hall, Tokyo (2012).
- Yanagida, M. Present, Past and Future of Life Science in Japan, RIKEN, Saitama (2012).
- Yanagida, M. In Okinawa, Japan and World, how young people live? A suggestion from a lifescientist, The Junior and Senior High School Affiliated to Showa Pharmaceutical University, Okinawa (2013).
- Yanagida, M. Thinking about my 50 years of research, Foundation for Biomedical Research and Innovation, Kobe (2013).
5. Intellectual Property Rights and Other Specific Achievements
Nothing to report
6. Meetings and Events
6.1 Seminar
- Title: Title: A tale of two condensins: universal organizers of chromosomes with diverse functions
- Date: February 22, 2013
- Venue: OIST Campus Lab1
- Speaker: Dr. Tatsuya Hirano (RIKEN Advanced Science Institute)
6.2 Seminar
- Title: How is genome DNA shaped into condensed chromosomes?: Opposing roles of transcription and condensin complex.
- Date: March 1, 2013
- Venue: OIST Campus Lab1
- Speaker: Dr. Takashi Sutani (Institute of Molecular and Cellular Bioscieneces, The University of Tokyo)