

FY2011 Annual Report
Cellular and Molecular Synaptic Function Unit
Professor Tomoyuki Takahashi

Abstract
In the neuronal system, dynamic changes of synaptic strength play critical roles in switching functional neuronal circuits. Regarding synaptic strength, compared with postsynaptic mechanisms much less is known for presynaptic mechanisms, primarily because of small nerve terminal structures preventing applications of electrophysiological and imaging techniques. The calyx of Held is a giant glutamatergic nerve terminal visually identified in the mammalian auditory brainstem slices. This fast relay synapse undergoes dramatic developmental changes in its structure, functional properties and molecular compositions during the second postnatal (P) week, when rodents start to hear sound at P10-12. By applying molecular, imaging and patch-clamp techniques to this synapse of developing rodents, we aim at elucidating presynaptic regulatory mechanisms underlying synaptic transmission. Our progress in the fiscal year 2011 is as follows:
(1) Discovery of an exo-endocytic coupling mechanism of synaptic vesicles.
After exocytic release of neurotransmitter, synaptic vesicles are retrieved by endocytosis and reused for another round of transmission. In order to maintain synaptic transmission at high frequency for a long period, exocytosis and endocytosis of vesicles must be balanced to prevent releasable vesicles from depletion and presynaptic terminals from abnormal swelling. We found that glutamate triggers NO production in the postsynaptic cell, and this NO retrogradely activate G-kinase and then PIP2 in the presynaptic terminal, thereby accelerates the rate of vesicle endocytosis. In fact, a PKG inhibitor directly loaded into a presynaptic terminal distorted the fidelity of synaptic transmission at 100 Hz after 30s.
(2) In situ determination of the rate at which synaptic vesicles are refilled with the neurotransmitter glutamate.
1. Staff
- Hiroshi Takagi, Group Leader
- Kohgaku Eguchi, Researcher
- Tetsuya Hori, Researcher
- Yukihiro Nakamura, Researcher
- Laurent Guillaud, Researcher
- Setsuko Nakanishi, Researcher
- Zacharie Taoufiq, Researcher
- Dimitar Dimitrov, Technician
- Kaori Egashira, Research Administrator
2. Collaborations
- Theme: Regulatory mechanisms for transmitter release
- Type of collaboration: Joint research
- Researchers:
- Professor, Naoto Saitoh, Doshisha University
- Takafumi Miki, Doshisha University
- Theme: Developmental changes in the presynaptic calcium transient profiles associated with transmitter release
- Type of collaboration: Scientific Collaboration
- Researchers:
- Professor, David DiGregorio, Pasteur Institute
- Theme: Developmental changes in the presynaptic calcium channels
- Type of collaboration: Scientific Collaboration
- Researchers:
- Professor, Ryuichi Shigemoto, National Institute for Physiological Sciences, Department of Cerebral Research, Division of Cellular Structure
- Ko Matsui, National Institute for Physiological Sciences, Department of Cerebral Research, Division of Cellular Structure
3. Activities and Findings
3.1 Maturation of a PKG-dependent retrograde mechanism for exo-endocytic coupling of synaptic vesicles (Eguchi et al, Neuron, 2012, in press).
In the presynaptic terminal, upon arrival of an action potential, neurotransmitter is released from synaptic vesicles by exocytosis, with vesicle membrane fusing with the terminal membrane. Vesicle membrane is then retrieved by endocytosis from the terminal membrane, refilled with neurotransmitter and reused for another round of neurotransmission. To maintain a stable neurotransmission the magnitude of exocytosis must somehow be informed to the machinery of endocytosis to keep balance between vesicle and terminal membrane because disruption of this balance can deplete intra-terminal vesicles, resulting in the failure of neurotransmission. Outbalance of exocytosis and endocytosis also distorts presynaptic terminal structure. Using membrane capacitance measurements at the calyx of Held presynaptic terminal we elucidated a molecular mechanism underlying exo-endocytic balancing. While investigating roles of protein kinases in the presynaptic terminal, we found that protein kinase G (PKG) inhibitors, when injected into the terminal, slowed the time course of vesicle endocytosis without affecting exocytosis or presynaptic Ca2+ currents (Fig 1).
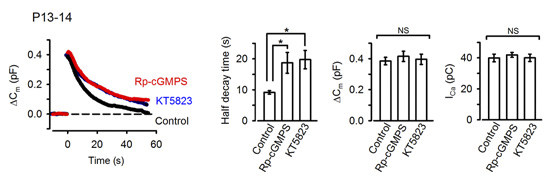
[Fig 1]
These pharmacological results suggest that PKG normally up-regulates the rate of endocytosis. Consistently, the PKG activating second messenger cGMP, when loaded into the calyx terminal, accelerated the endocytic half-time, significantly at above 300 mM (Fig 2).
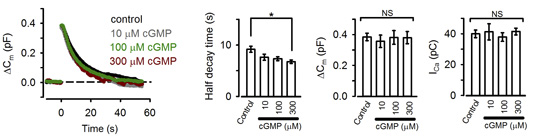
[Fig 2]
NO is known to increase intracellular cGMP by stimulating guanyryl cyclase. We tested whether NO is linked to presynaptic PKG. The NO scavenger PTIO, when bath-applied, mimicked the PKG inhibitors in the slowing effect on endocytosis. Furthermore, in the presence of PTIO, the PKG inhibitor Rp-cGMPS no longer affected endocytic time course (Fig 3).
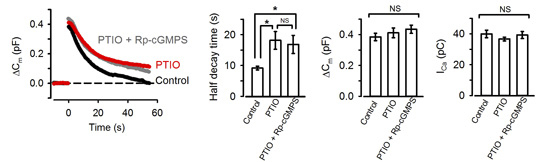
[Fig3]
Namely PTIO occluded PKG in the slowing effect of endocytosis. These results also imply that NO present in the extracellular medium permeates presynaptic terminal membrane, thereby upregulating PKG activity. Where does this NO come from then?
Neurotransmitter glutamate, when released from calyceal terminals by exocytosis, activates postsynaptic Ca2+ permeable glutamate receptors, such as NMDA receptors, thereby increasing intracellular Ca2+ concentration. Because Ca2+ can trigger NO synthesis by activating Ca2+-dependent NO synthetase, we tested whether NMDA receptors might reside in the upstream cascade of PKG. As illustrated in Fig 4, bath-application of the NMDA receptor antagonist D-AP5 mimicked and occluded the effect of the PKG inhibitor.
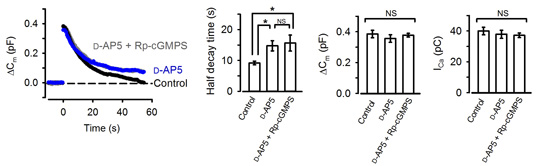
[Fig 4]
These results suggest that activation of postsynaptic NMDA receptors by the neurotransmitter glutamate normally accelerate vesicle endocytosis. In this way, the magnitude of exocytosis is reported to the machinery of endocytosis, thereby balancing the exo-endocytosis. Given that the experiments are performed at a single calyx-MNTB synapse with one to one innervation, the message of the amount of neurotransmitter released must flow from a postsynaptic MNTB neuron to a presynaptic calyx terminal.
This retrograde signaling mechanism reports the magnitude of exocytosis from postsynaptic neuron to presynaptic endocytic machinery, thereby regulating the rate of vesicle endocytosis for exo-endocytic balance (Fig 5).
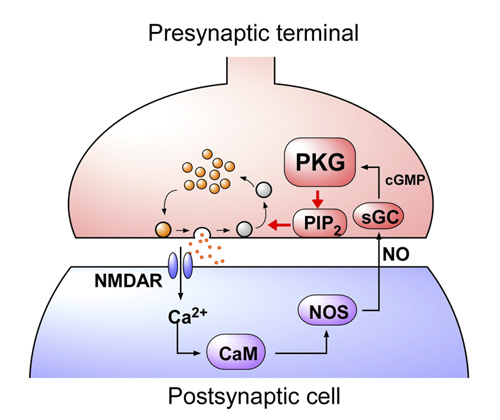
[Fig 5]
What happens to neurotransmission if this feedback mechanism is inhibited? To address this question, we monitored postsynaptic action potentials elicited by presynaptic stimulation at 100 Hz (Fig 6A). In slices, at room temperature, presynaptic action potentials elicited continuously at 100 Hz were followed by postsynaptic action potentials with a failure of less than 30 % in 50s (Fig 6B, C). When a PKG inhibitor was loaded into the calyx presynaptic terminal, the number of failures of postsynaptic action potentials markedly increased and became >50% in 50s. These results suggest that the PKG-dependent exo-endocytic coupling mechanism is indispensable for the maintenance of synaptic transmission.
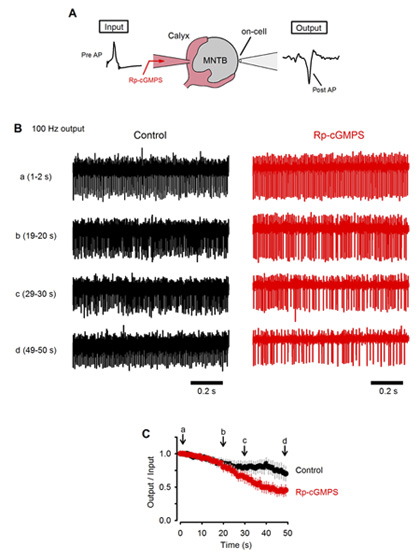
[Fig 6]
The calyx of Held undergoes remarkable development during the second postnatal week when rodents start to hear sounds. As the above results were obtained from postnatal day (P) 13-15 rats, we next asked whether the exo-endocytic coupling mechanism might operate before hearing onset. At P7-9 prehearing terminal, endocytosis was significantly slower than that at P13-15, and the PKG inhibitor had no effect on the time course of endocytosis (Fig 7). Interestingly, the endocytic half-time at P7-9 calyces was similar to that at P13-14 calyces loaded with PKG inhibitors, suggesting that the maturation of the PKG-dependent exo-endocytic coupling system underlie the developmental speeding of the endocytosis.
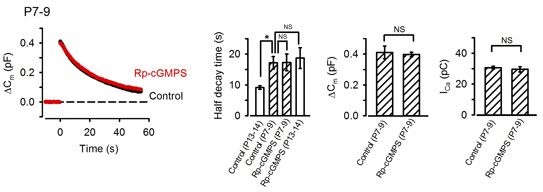
[Fig 7]
Is PKG expressed in presynaptic terminal before hearing onset? Immunocytochemical and western blot analysis clearly indicate that PKG expression in the calyx terminal as well as in the whole brainstem is low before hearing onset, but it is significantly up-regulated during the second postnatal week (Fig 8). Thus, the PKG-dependent exo-endocytic coupling mechanism is developed as animals mature, thereby contributing the establishment of the high-fidelity high frequency neurotransmission at this fast synapse. Importantly, a similar mechanism has been reported at hippocampal synapses in culture (Micheva et al, 2003), suggesting that this PKG-dependent exo-endocytic coupling mechanism may operate at many type of central synapses.
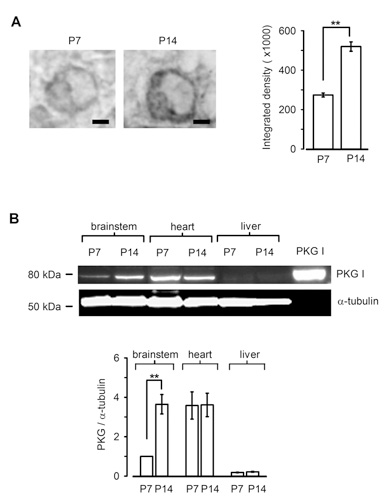
[Fig 8]
The remaining question on the signal mechanism is how PKG accelerate vesicle endocytosis. Among molecules involved in vesicle endocytosis, phosphoinositides PIP2 plays a major role in clathrin-mediated endocytosis (CME). We found that the PIP2 inhibitor PAO, when injected into the calyx terminal, mimicked and occluded the effect of PKG (Fig 9A) with no effect on exocytosis or presynaptic Ca2+ currents. Furthermore, the inhibitory effect of PAO could be counteracted by additional injection of PIP2 (Fig 9B).
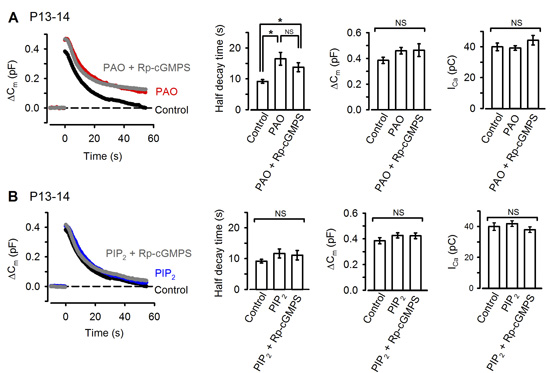
[Fig 9]
Immunocytochemical examination revealed clear PIP2 immunoreactivity (IR) in the calyx of Held (Fig 10A). Treatment of slices with the PKG inhibitor or PTIO significantly diminished the PIP2 IR in the terminal (Fig 10A, B). Consistently Elisa assay indicated that PIP2 content in the brainstem tissue declined after treatments with the PKG inhibitor or PTIO, whereas these treatments had no effect for brainstem tissues from P7-8 rats (Fig 10C). Taken together, these results suggest that PIP2 resides in the downstream cascade of PKG and regulate the rate of vesicle endocytosis.
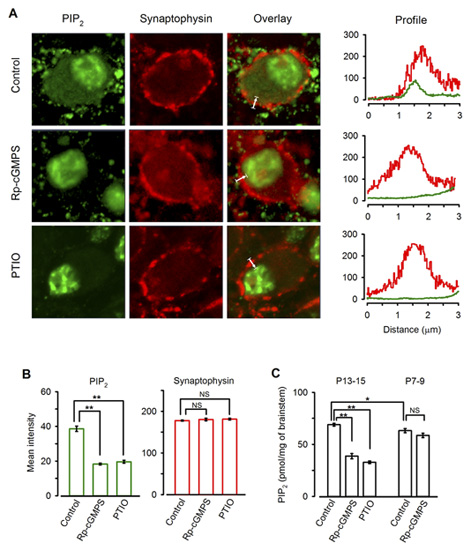
[Fig 10]
These findings have opened up a question as to the molecules, which link PKG and PIP2. We are currently looking for the answer to this question.
3.2 Synaptic vesicle filling with neurotransmitter as a rate-limiting step for vesicle reuse (Hori et al, manuscript under review)
After exocytic release of transmitter, synaptic vesicles are retrieved by endocytosis and recycled to be reused. At glutamatergic excitatory synappses imaging studies using fluorescence indicators have revealed different types of vesicle endocytosis with different kinetics, ranging from subsecond to tens of seconds. It has been argued that fast endocytosis and recycling are required for small synapses with limited number of releasable vesicles. Despite the wealth of knowledge on the endocytic kinetics, little is known for the time required for vesicles to be refilled with neurotransmitter. This information is important because empty vesicles do not contribute to neurotransmission. In isolated or reconstructed vesicles, the uptake time constant of the neurotransmitter glutamate goes beyond several minutes. This time course is far too slow to account for the recovery of synaptic transmission after depletion of releasable vesicles. Clearly, the original kinetics of glutamate uptake into vesicles is distorted during the procedure of vesicle isolation or reconstruction.
It is therefore desirable if one can measure the time of vesicular glutamate uptake in situ. We challenged this problem using the calyx of Held presynaptic terminal. In this terminal, whole cell wash out of cytosolic glutamate leads to a depletion of glutamate in vesicles, because of vesicle recycling combined with a passive leakage. Glutamate depletion from vesicles results in a reduction of the frequency and mean amplitude of spontaneous miniature, i.e., vesicular, EPSCs and mean amplitude of nerve-evoked EPSCs (Ishikawa et al, 2002 Neuron). In contrast, injection of glutamate into the terminal increases vesicular glutamate content and the size of EPSCs. To address the question on the kinetics of vesicle refilling with glutamate, we introduced the caged glutamate compound MNI-glutamate (2-10 mM) into the terminal.
When glutamate was washed out from the terminal, the EPSC amplitude declined to a low level (<20%). Application of an UV flash light (1s) caused a recovery of EPSCs to the original amplitude (>95%). This recovery was completely abolished in the presence of bafilomycin in the bath solution. Bafilomycin blocks acidification of vesicles by vacuolar type H+-ATPase that is required for glutamate uptake with vesicular glutamate transporters (VGLUTs). This recovery was not associated with changes in the membrane capacitance, suggesting that glutamate uncaging had no effect on exocytosis of vesicles. Furthermore, glutamate uncaging increased the frequency and amplitude of miniature EPSCs, suggesting that vesicles were refilled with glutamate photo-released from the MNI compound.
From these data we analyzed the kinetics of glutamate uptake. To gain information as to the glutamate concentration raised by glutamate uncaging, we loaded given concentration of glutamate into calyces and measured the EPSC amplitude. We then uncaged glutamate from the MNI compound at different concentrations (2-10 mM). By normalizing the maximal EPSC amplitude between glutamate loading and MNI-glutamate uncaging, we obtained the relationship between the time constant of EPSC recovery and glutamate concentrations. Lineweaver-Burk plot analysis of these data indicated that the maximal glutamate uptake rate (Vmax) was 15 sec and the glutamate affinity of the transporter (Km) was 0.9 mM. The latter value is similar to what have been reported for VGLUT1 and VGLUT2.
The 15s time constant measured for glutamate refilling into vesicles is fast enough to fill up vesicles in the CME pathway, but it is too slow to fill vesicles undergoing fast recycling pathways such as the “kiss-and-run” fusion flicker that is thought to have subsecond time constant. In order to contribute to synaptic transmission, vesicles after recycling through fast pathways must wait for them refilled with transmitter before reused. In fact, it has been estimated that “kiss-and run” vesicles are reused only after 23 sec. This time course is not much different from that of CME. Thus, it remains to be re-examined whether the fast recycling mechanisms have physiological significance.
In isolated vesicles, glutamate uptake is known to have a biphasic dependence of Cl- concentrations. Presynaptic terminals including the calyx of Held possess Cl- -permeable channels such as glycine receptors, therefore intra-terminal Cl- concentration can change from time to time. We tested whether glutamate refilling depends upon Cl- at the calyx of Held. When [Cl-] was reduced below mM, the rate and magnitude of glutamate refilling was markedly reduced. When [Cl-] was increased above 100 mM, the rate of glutamate refilling was significantly slowed. Thus, these results in situ agree with those previously reported with isolated vesicles.
If vesicle refilling time limits the time for vesicle reuse, manipulations to retard the refilling rate will cause defects in neurotransmission. We are currently studying this issue.
4. Publications
4.1 Journals
4.2 Books and other one-time publications
4.3 Oral and Poster Presentations
1. Hori, T. Refilling rate of synaptic vesicles with glutamate in the mammalian presynaptic terminal, OIST workshop: Molecular & Structural Organization of Presynaptic Function and Plasticity, OIST, Okinawa, Japan, September 7, 2011
5. Intellectual Property Rights and Other Specific Achievements
Nothing to report
6. Meetings and Events
International Workshop "Molecular & Structural Organization of Presynaptic Function and Plasticity"
- Date: Sep. 7-9, 2011
- Venue: OIST Campus
- Co-organizers:
- Ian D Forsythe (University of Leicester)
- Shigeo Takamori (Doshisha University)
- Speakers:
- Dieter Bruns (University of Saarland, Germany)
- Yukiko Goda (Riken Brain Science Institute)
- Volker Haucke (Free University of Berlin)
- Reinhard Jahn (Max Planck Institute for Biophysical Chemistry, Germany)
- Alan Marty (Paris Descartes University)
- Christopher J McBain (National Institutes of Health, USA)
- Erwin Neher (Max Planck Institute for Biophysical Chemistry, Germany)
- Silvio Rizzoli (European Neuroscience Institute, Germany)
- Tim A Ryan (Weill Cornell Medical College, USA)
- Ryuichi Shigemoto (National Institute for Physiological Sciences, Okazaki)
- Oleg Shupliakov (Karolinska Institute, Sweden)
- Jakob Soerensen (University of Copenhagen)
- Lucia Tabares (University of Seville School of Medicine, Spain)
- Ian D Forsythe (University of Leicester, UK)
- Shigeo Takamori (Doshisha University)
- Henrique von Gersdorff (Oregon Health & Science University)
7. Other
Nothing to report.