

FY2023 Annual Report
Chemistry and Chemical Bioengineering Unit
Professor Dr. Fujie Tanaka, Professor
Abstract
The ability to design and synthesize organic molecules constitutes the foundation that underlies basic research as well as applied science. The ability is also essential for the development of pharmaceuticals and biofunctional molecules. This unit develops efficient, concise, and safe chemical transformation methods and strategies for constructing small molecules bearing functional groups and/or chiral centers that are relevant to the creation of biofunctional molecules. We design and create small organic molecule catalyst systems for designed chemical transformations, and we develop reaction strategies that use small organic molecules as enzyme-like catalysts. With the use of organic molecules as catalysts, we minimize the need for protection and deprotection steps that are usually required for the synthesis of functionalized molecules. When a reaction method does not affect functional groups that are not at the reaction site, the reaction method can be used for the synthesis of a series of molecules bearing various functional groups. This means that a series of molecules of interest may be synthesized using the same method in a short route, providing advantages for the synthesis of biofunctional candidate molecules. We also investigate the chemical bases of the reactions to understand the mechanisms of the catalysis and molecular interactions provided by organic molecules. By taking advantage of the use of features of our developing molecules, we also develop strategies and methods for conjugation of proteins and peptides with other molecules. The research undertaken by this unit advances the chemistry of catalysis and of molecular synthesis. The studies by this unit accelerate the creation of molecules used in biomedical research and contribute to the development of new therapeutics, therapeutic strategies, and diagnostic methods.
1. Staff
- Dr. Venkati Bethi
- Dr. Santosh Chavan
- Dr. Yuvraj Garg
- Dr. Akash Jana
- Dr. Kamal
- Dr. Nandkishor Khot
- Dr. Bapurao Dattatray Rupanawar
- Dr. Muhammad Sohail
- Dr. Prakash K. Warghude
- Santanu Mondal, Graduate Student
- James Osborne, Graduate Student
- Kuan-Lin Chen, Graduate Student
- Anjali Gupta, Graduate Student
- Daria Sherstiukova, Graduate Student (Rotation)
- Shiho Chinen, Research Unit Administrator
2. Activities and Findings
2.1. Development of catalysts and catalytic chemical transformation methods for the synthesis of functionalized molecules
We have been developing small organic molecule catalysts (organocatalysts) and organocatalytic molecular transformation methods useful for the concise synthesis of functionalized molecules under mild conditions. We have also been investigating the mechanisms of these catalyzed reactions and key factors that control the catalyses and the selectivities in the chemical transformations. Through our studies, we will further our understanding of the chemistry of catalyses, molecular interactions, and chemical transformations.
2.1.1. Design and synthesis of 1,3-diamine-derived catalysts and the use in Mannich reactions
Compounds that have a primary amine group and a tertiary amine group in a geminal relationship (or 1,2-relationship), such as cinchona-derived amines, cyclohexane-1,2-diamines, and 1,2-diarylethane-1,2-diamines, have been used as catalysts with or without other molecules (such as acids) to accelerate bond-forming reactions of enolizable ketones and aldehydes as nucleophiles. During the catalysis by these amines, the primary amine often forms an enamine intermediate, and the tertiary amine group is protonated and functions as an acid to interact with electrophiles. When the tertiary amine group interacts with acid molecules, it may also provide a steric bulk and/or may have a shielding function. We reasoned that molecules bearing primary amine groups and tertiary amine groups in a 1,3-relationship would work as catalysts and that such catalysts would provide scope of the catalysis different from the 1,2-diamine-derived catalysts. Thus, we have designed ans evaluated 1,3-diamine-derived catalysts.
We have synthesized catalyst 1a (Scheme 1) (Garg, Osborne, Vasylevskyi, Velmurugan, and Tanaka, J. Org. Chem. 2023, 88, 11096) using our previously reported diastereo- and enantioselective Mannich reaction catalyzed by β-proline in the presence of potassium carbonate (Garg and Tanaka, Org. Lett. 2020, 22, 4542). We have demonstrated that 1a catalyzes enantioselective Mannich reactions of ketones in the presence of acids (Garg, Osborne, Vasylevskyi, Velmurugan, and Tanaka, J. Org. Chem. 2023, 88, 11096). Importantly, catalyst 1a is useful for catalyzing enantioselective γ-position-selective Mannich reactions of β-ketophosphonates (Scheme 2) (Garg, Osborne, Vasylevskyi, Velmurugan, and Tanaka, J. Org. Chem. 2023, 88, 11096). We have also synthesized catalysts 1b-g (Figure 1) (Garg, Osborne, Vasylevskyi, Velmurugan, and Tanaka, J. Org. Chem. 2023, 88, 11096). Evaluations of these catalysts indicated that the 1,3-diamine moiety of 1 is necessary for the catalysis of the Mannich reaction. When the Mannich reaction in the presence of 1a•CF3COOH with dibenzyl phosphate was analyzed at various time points, no formation of α-position reaction product was detected at any time point analyzed, and the enantiopurity of the γ-position reaction product was essentially unchanged (er 95:5-96:4) during the reaction. Thus, the C-C bond formation selectively occurred at the γ-position of the β-ketophosphonate, and the Mannich reaction product stereochemistry was controlled at the C-C bond-formation step.
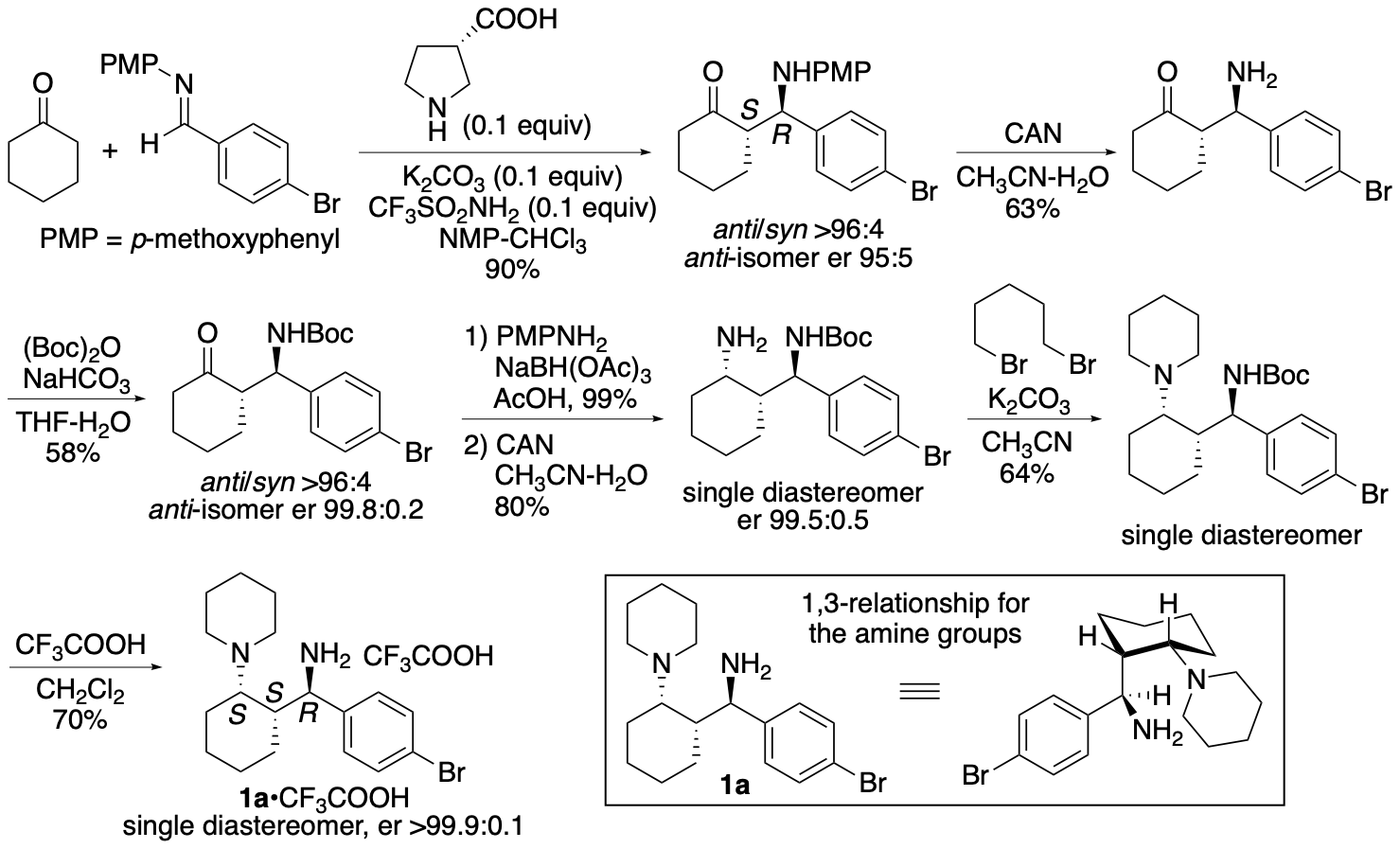
Scheme 1
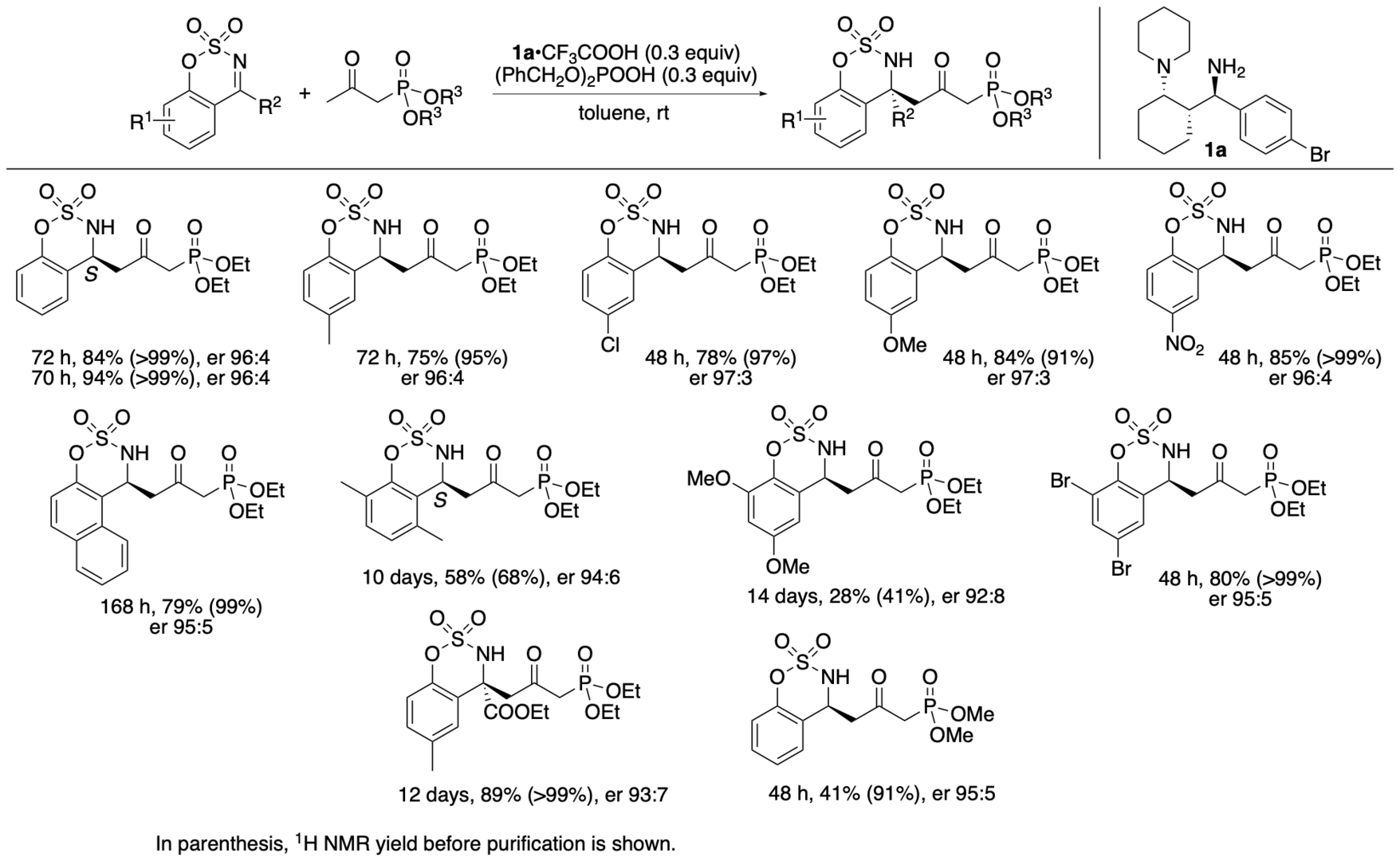
Scheme 2
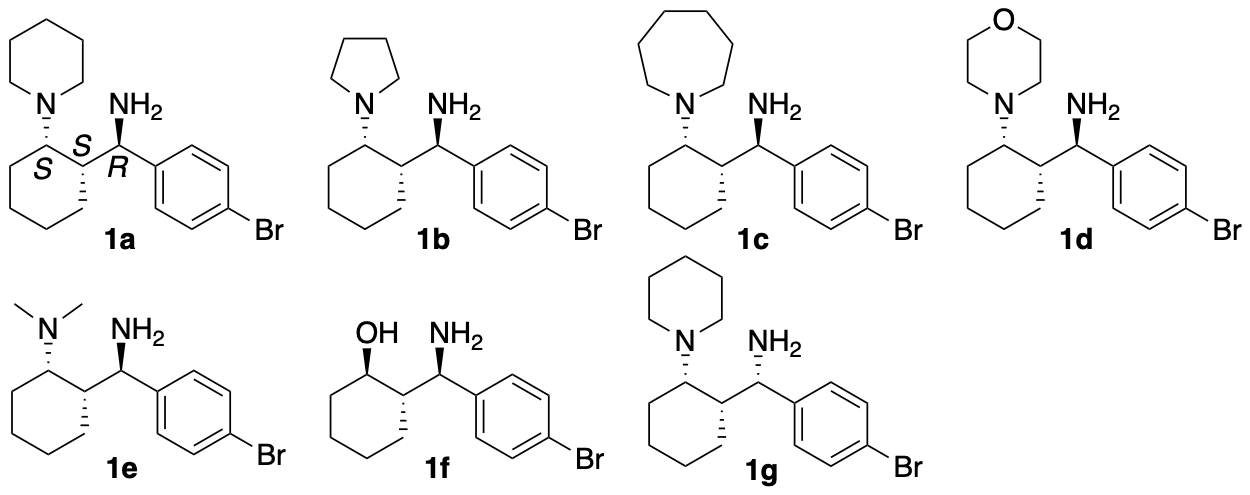
Figure 1
The catalyst system with 1a also catalyzes Mannich reactions of other ketones (Scheme 3) (Garg, Osborne, Vasylevskyi, Velmurugan, and Tanaka, J. Org. Chem. 2023, 88, 11096). With the exception of the reaction of ethyl acetoacetate, in which the regioisomer was also formed, the bond formation occurred enantioselectively at the less substituted α-position of the ketone carbonyl group (or at the methyl group).
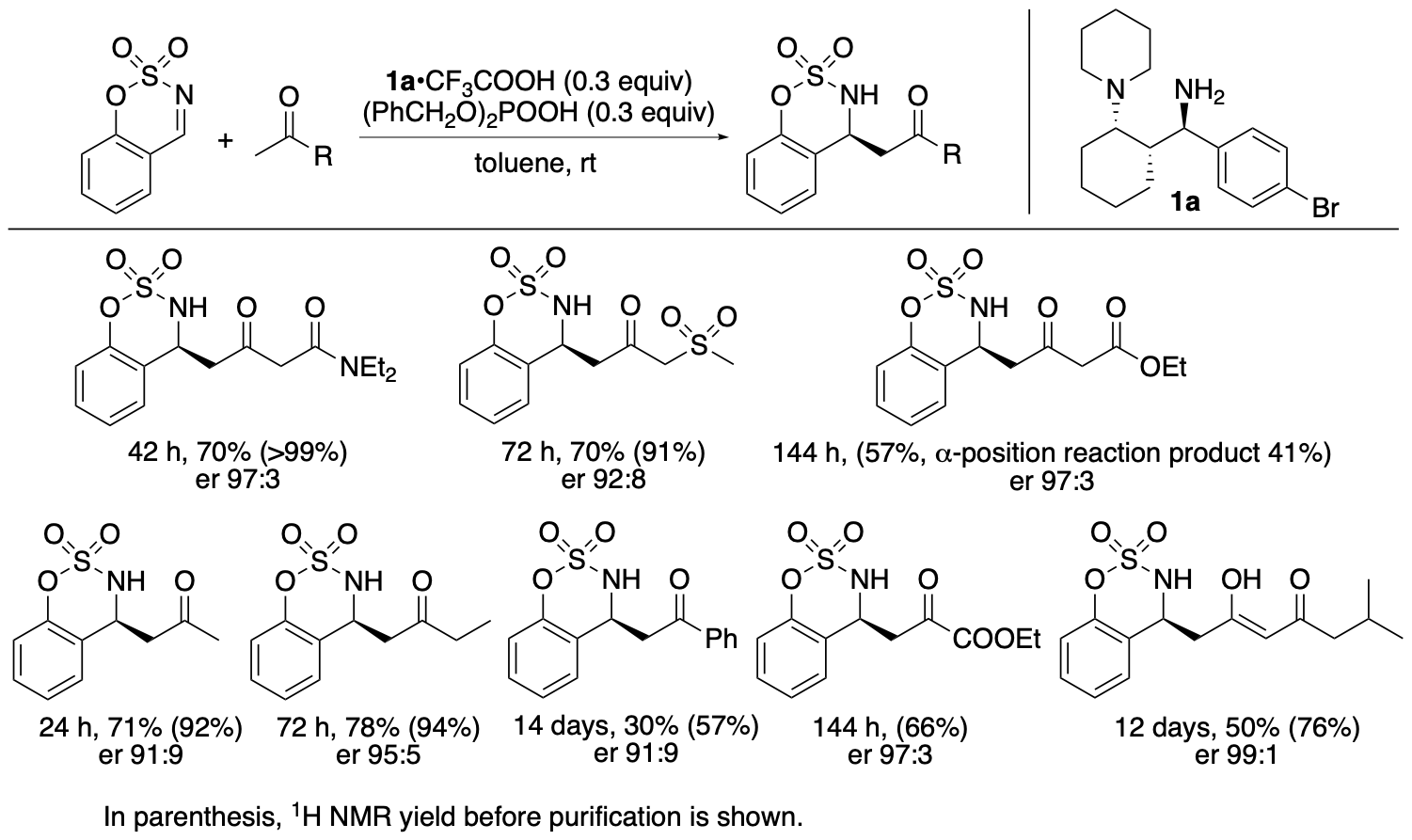
Scheme 3
We previously developed enantioselective γ-position selective Mannich reactions of β-ketoesters (such as ethyl acetoacetate) catalyzed by cinchona-derived amine and acids (Bethi and Tanaka, Org. Lett. 2022, 24, 6711). The catalyst system composed of 1,3-diamine derivative 1a and acids resulted in the formation of a mixture of α- and γ-position reaction products in the Mannich reaction of ethyl acetoacetate. For Mannich reactions of β-ketophosphonates, the 1a catalyst system catalyzed the γ-position-selective Mannich reaction; the α-position reaction product was not formed. The catalytic features and the scopes of the reactions catalyzed by 1,3-diamine-derived catalyst 1a are unique.
Our development of the 1,3-diamine-derived catalysts and the investigation of the reactions catalyzed by the 1,3-diamine-derived catalysts provided the information about the relationships between catalyst structures and the catalytic features and mechanisms. We are continuing the development of catalysts and catalytic chemical transformations.
2.1.2. Selective vinylogous aldol reactions of β-methyl-substituted cyclic enones with isatins catalyzed by DBU and pyrrolidine to afford 3-substituted 3-hydroxyoxindoles
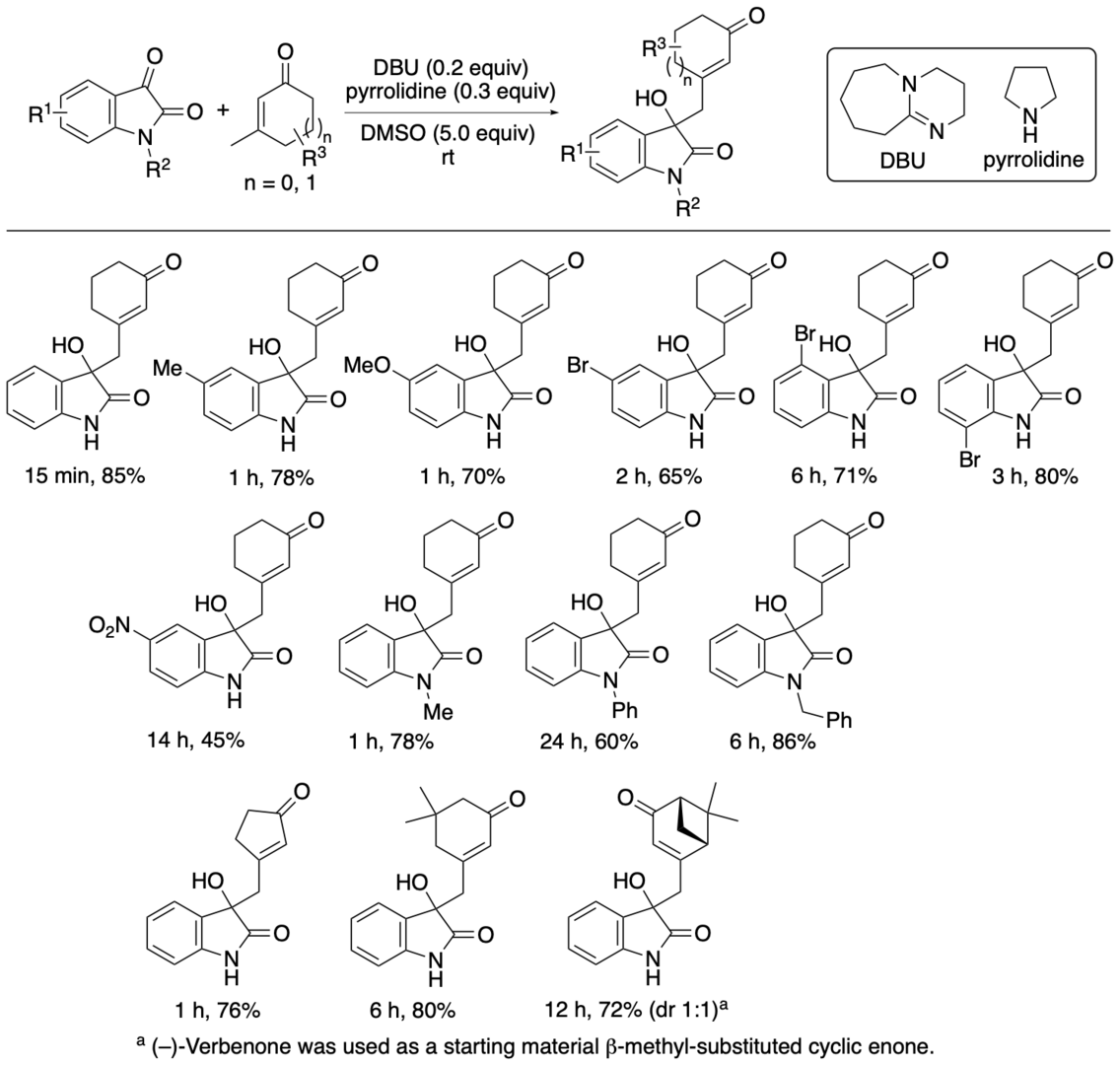
3-Substituted 3-hydroxyoxindoles are found in bioactive natural products. The 3-substituted 3-hydroxyoxindoles have often been synthesized by aldol reactions of isatins and carbonyl derivatives, in which the latter acts as a nucleophile (e.g., enolate, enol derivative, enamine). In these reported aldol reactions, the bond formation usually occurs at the α-position of the carbonyl group, although the bond formation can occur at other positions depending on carbonyl derivatives. We have developed aldol reactions of isatins with β-methyl-substituted cyclic enone derivatives, such as 3-methyl-2-cyclohexen-1-one, catalyzed by DBU-pyrrolidine (1:1.5) that afford aldol products with the bond formation at the methyl group (or at the one of the two g-positions) of the β-methyl-substituted cyclic enone derivatives (Scheme 4) (Bethi, Bakhtawar, Rupanawar, and Tanaka, Asian J. Org. Chem. 2023, 12, e202300193).
Scheme 4
In the reaction of isatin and 3-methyl-2-cyclohexen-1-one, the use of DABCO or Et3N rather than DBU in the DBU-pyrrolidine (1:1.5) catalyst system resulted in no formation of the aldol products. Pyrrolidine alone did not catalyze the reaction: When the reaction was performed in the presence of pyrrolidine and DMSO without DBU, no formation of the aldol products was observed. DBU alone catalyzed the reaction but the product regioselectivity was moderate (2:1). Addition of pyrrolidine to the DBU catalysis in the presence of DMSO enhanced the reaction rate more than 10-fold and improved the regioselectivity to afford the product as almost a single regioisomer, in which the bond formation occurred at the methyl group of the 3-methyl-2-cyclohexen-1-one. Thus, whereas DBU is essential for the catalysis, both pyrrolidine and DMSO are important for the enhanced reaction rate and for the formation of the product with almost perfect regioselectivity.
When the formation of the products in the reaction of isatin and 3-methyl-2-cyclohexen-1-one in the presence of DBU, pyrrolidine, and DMSO was analyzed at various time points, essentially only the product with the bond formation at the methyl group was observed, from the initial stage of the reaction at 1 min (40% conversion) to the stage of the completion of the reaction at 15 min (100% conversion) and even after 1 h. The results of our mechanistic investigation suggested that the aldol product with the bond formation at the methyl group of 3-methyl-2-cyclohexen-1-one was selectively formed directly from the starting materials.
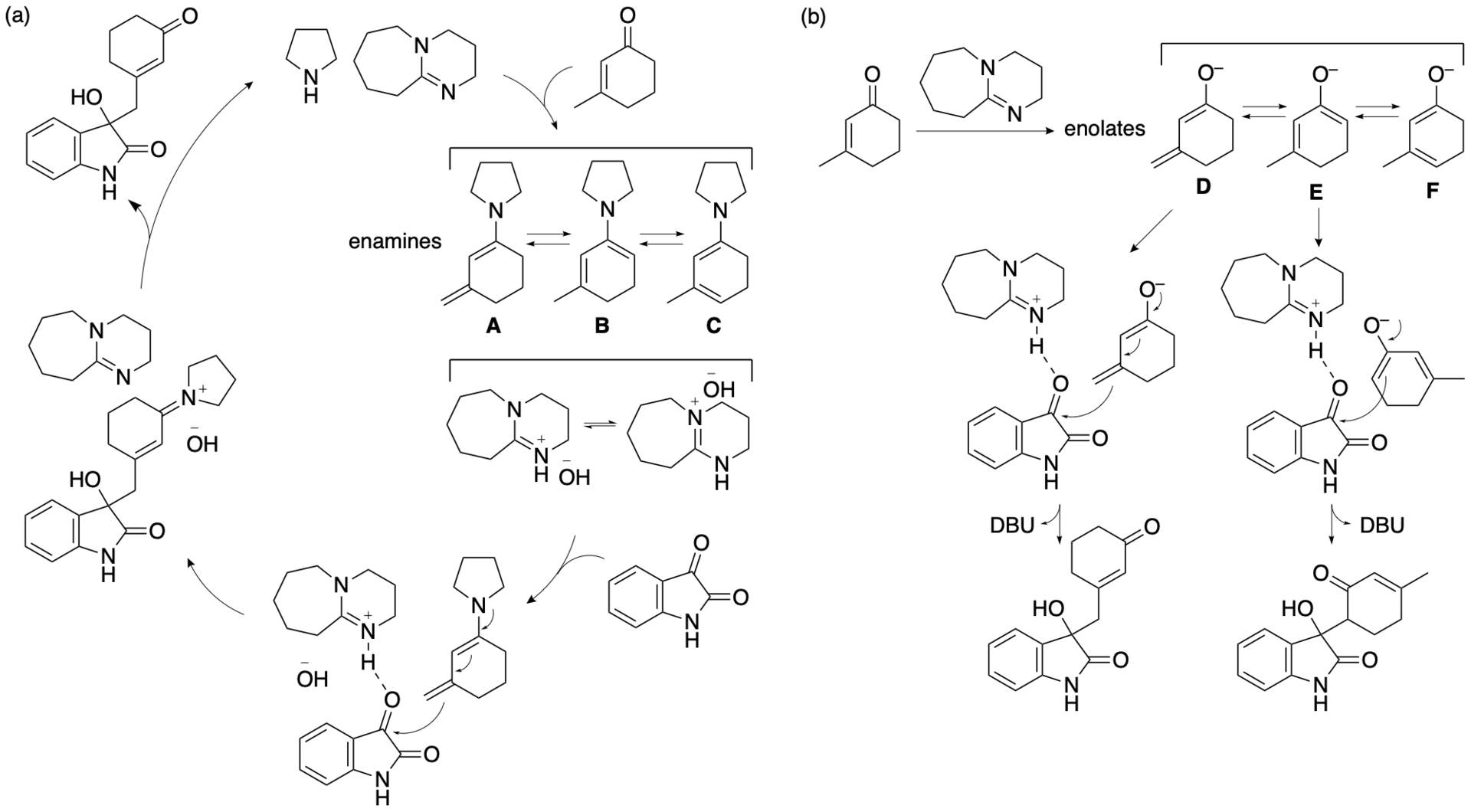
Based on the results, we have proposed plausible mechanisms for the formation of the product (Bethi, Bakhtawar, Rupanawar, and Tanaka, Asian J. Org. Chem. 2023, 12, e202300193). In the reaction of isatin and 3-methyl-2-cyclohexen-1-one in the presence of DBU, pyrrolidine, and DMSO, enamines A-C may be formed (Scheme 5a). Among the formed enamines, enamine A would lead to the formation of the product. Because a ratio of pyrrolidine to DBU of 1.5:1 resulted in the formation of the γ-position reaction product (i.e., the vinylogous aldol reaction product) with almost perfect selectivity but a ratio of 1:1 resulted in lower regioselectivity, we reasoned that the use of less pyrrolidine or the use of DBU in the absence of pyrrolidine results in the formation of enolates D, E, and F and that enolate E leads to the formation of the α-position reaction product (Scheme 5b). Thus, the formation of the enamines appears to be key for the selective bond formation at the methyl group of 3-methyl-2-cyclohexen-1-one. It is likely that the formation of enamine A requires both pyrrolidine and DBU and that DMSO may enhance the formation of the enamines.
During the catalysis, DBU acts as a base to abstract protons, and the protonated DBU may act as an acid to interact with the carbonyl group to accelerate the C-C bond formation. DMSO may also stabilize the charged transition state. Among the formed enamines, enamine A may be the most reactive based on the stereoelectronic effect. DMSO may efficiently stabilize the charged transition state of the C-C bond formation involving enamine A. In other words, the transition state leading to the C-C bond formation at the methyl group of 3-methyl-2-cyclohexen-1-one that is generated from enamine A may be the most stable of the transition states generated from enamines A, B, and C. Populations of the enamines formed may not be related to the stabilities of the transition states generated leading to the bond formations.
Scheme 5
Protonated DBU can function as an acid under the basic conditions in the presence of DBU and pyrrolidine. However, DABCO and Et3N may not act as acids in the presence of pyrrolidine in DMSO. That DBU can act as an acid after protonation may be key for the catalysis by the DBU-pyrrolidine. Because of the amidine moiety of the DBU, the exchange between the protonated and non-protonated forms may occur quickly, and the rapid dynamics may be responsible for efficient catalysis in the presence of pyrrolidine.
We have developed the catalyst system composed of DBU and pyrrolidine, a new type of combination catalyst system, which contains two types of amine derivatives. The use of the DBU-pyrrolidine catalyst system has enabled the vinylogous aldol reactions.
2.1.3. Enantioselective Mannich and retro-Mannich reactions and combinations of these reactions to afford tetrasubstituted α-amino acid derivatives
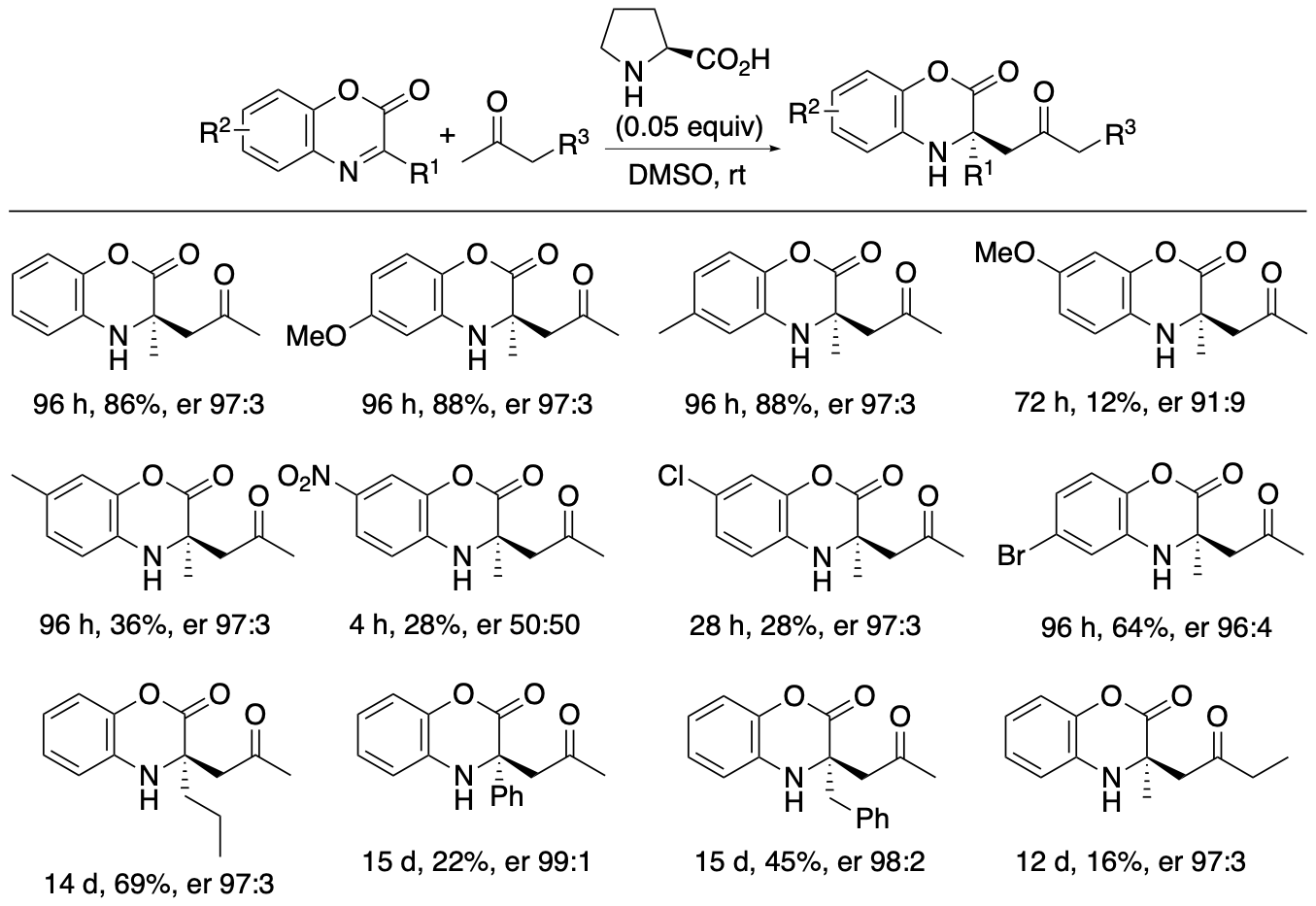
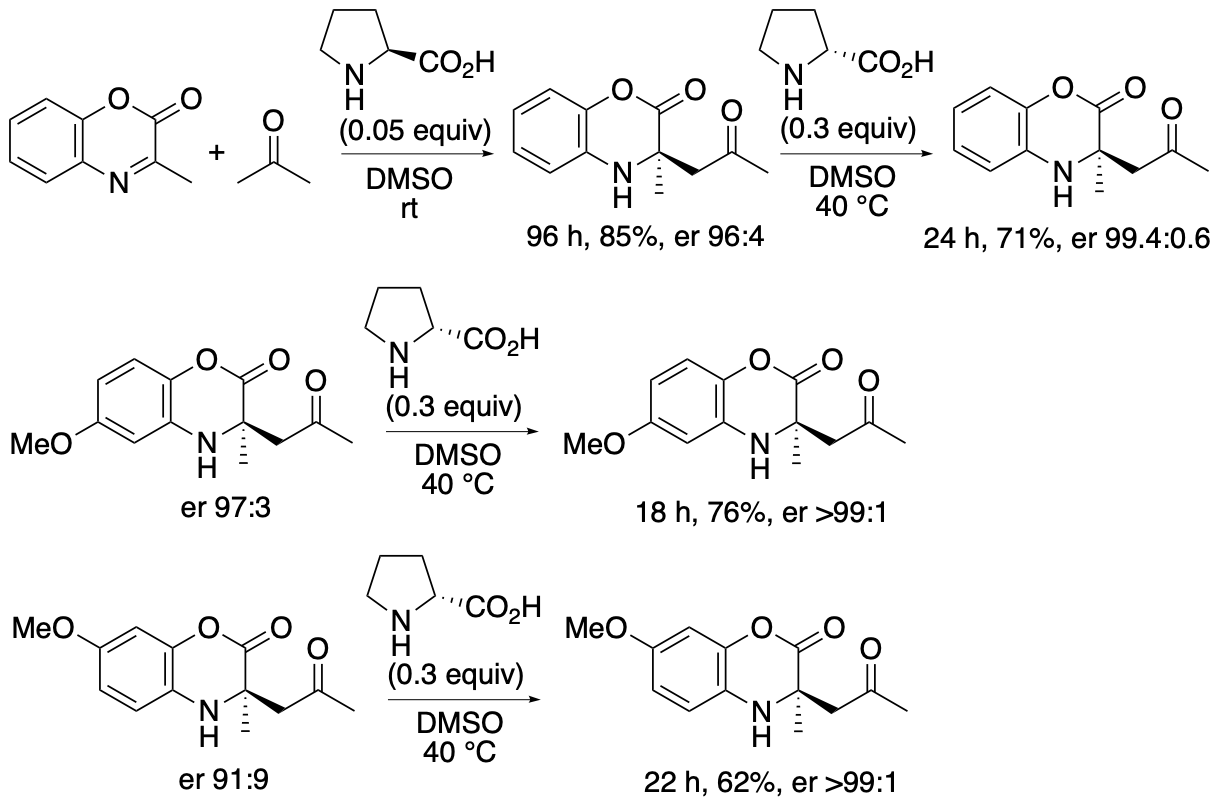
α,α-Disubstituted-a-amino acid derivatives or tetrasubstituted a-amino acid derivatives are found in pharmaceuticals and their building blocks. We have developed proline-catalyzed enantioselective Mannich reactions of 1,4-benzoxazinone-derived cyclic ketimino esters and ketones to afford tetrasubstituted α-amino acid derivatives (Scheme 6) (Chen and Tanaka, Org. Biomol. Chem. 2024, 22, 477). We have also developed kinetic resolutions of the Mannich reaction products via retro-Mannich reactions (Scheme 7) (Chen and Tanaka, Org. Biomol. Chem. 2024, 22, 477). Further we have developed a strategy that combines the Mannich reactions with the retro-Mannich reaction to access to highly enantiomerically enriched tetrasubstituted a-amino acid derivatives with er >99:1, which are difficult to obtain from either reaction alone (Chen and Tanaka, Org. Biomol. Chem. 2024, 22, 477).
Scheme 6
Scheme 7
The 1,4-benzoxazinone ring structure is often present in bioactive molecules. Thus, Mannich reaction products bearing the benzoxazinone moiety should be useful in drug discovery efforts. In addition, the benzoxazinone-derived cyclic amino esters can be transformed to other derivatives by reactions on the ester group and by the deprotection of the phenol group attached to the amine group. The use of cyclic ketimino esters avoids potential hydrolysis of the imine during Mannich reactions. Highly enantioselective Mannich reactions of ketimino esters in which a simple alkyl group (such as methyl group) is substituted on the imine carbon of the ketimino ester (i.e., the substituent is not electron-withdrawing) have rarely been reported. Thus, we have explored Mannich reactions of ketones with cyclic ketimino esters bearing alkyl and aryl substituents on the imine carbon.
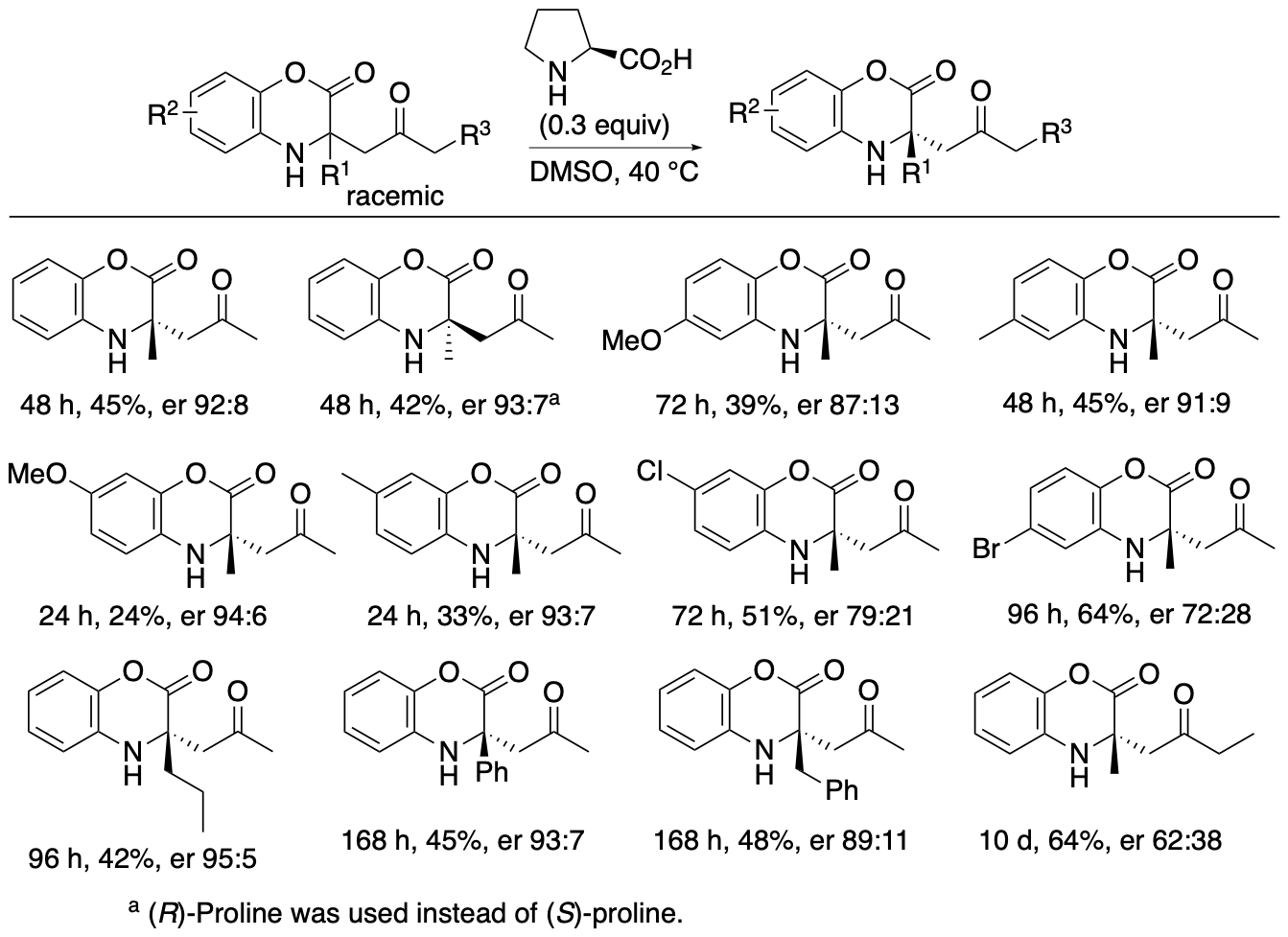
Both the Mannich reactions and the kinetic resolutions via retro-Mannich reactions afforded enantiomerically enriched products, but the enantiopurities of the products were not perfect. To obtain the products with er >99:1, the (S)-proline-catalyzed Mannich reaction and the (R)-proline-catalyzed retro-Mannich reaction were combined (Scheme 8). When Mannich products 3 formed by the Mannich reaction in the presence of (S)-proline were treated with (R)-proline, the minor enantiomers were decomposed, and products 3 with er >99:1 were obtained.
Scheme 8
2.1.4. Enantioselective reactions of pyruvates
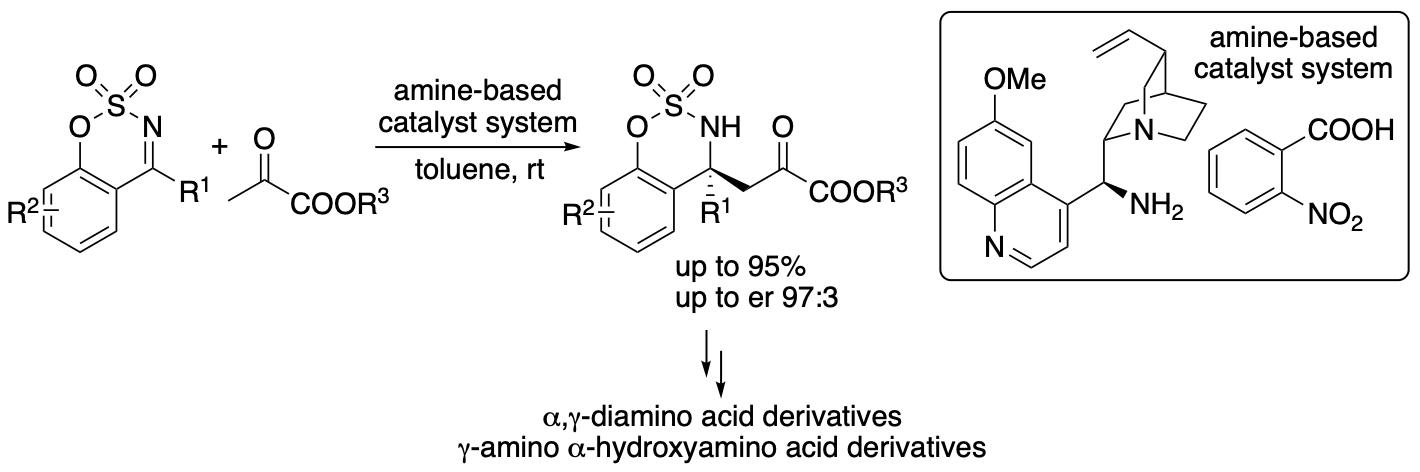
Pyruvates or 2-oxopropanoates act as electrophiles and nucleophiles and are useful compounds for the synthesis of various molecules. In Nature, pyruvate is a precursor for amino acids, sugars, cofactors, and other biofunctional molecules. In enzyme-catalyzed reactions, pyruvates often serve as nucleophiles. In contrast, in the majority of non-enzymatic reactions of pyruvates, pyruvates serve as electrophiles due to the activated reactivity of the α-ketocarbonyl structure. Without the use of enzymes, the control of the nucleophilic reactivities of pyruvates, especially simple pyruvates such as ethyl pyruvate, is difficult. Reactions of pyruvates often results in the formation of self-aldol reaction products and cascade reaction products, in which pyruvates act as both nucleophiles and electrophiles. We previously developed and reported direct enantioselective Mannich reactions of pyruvates with cyclic imines catalyzed by an amine-based catalyst system, in which pyruvates act as nucleophiles without acting as electrophiles (Scheme 9) (Mondal, Aher, Bethi, Lin, Taniguchi, Monde, and Tanaka, Org. Lett. 2022, 24, 1853). The acidity and the structure of the acid used in the amine-based catalyst system influenced the yield and the enantioselectivity; the acid component of the amine-based catalyst system was key to obtain the desired Mannich products in high yields with high enantioselectivities. In addition, using the α-ketoester group of the products as the reaction site, α,γ-diamino acid derivatives and γ-amino α-hydroxyacid derivatives were readily synthesized.
Scheme 9
To expand the strategies that enable the reactions of pyruvates as nucleophiles, we have been developing catalytic enantioselective Mannich reactions of substituted pyruvates (such as 2-oxobutanoates etc.) with cyclic and acyclic imines. We are also developing catalytic enantioselective aldol reactions of pyruvates.
2.1.5. Construction of spirooxindole polycyclic derivatives
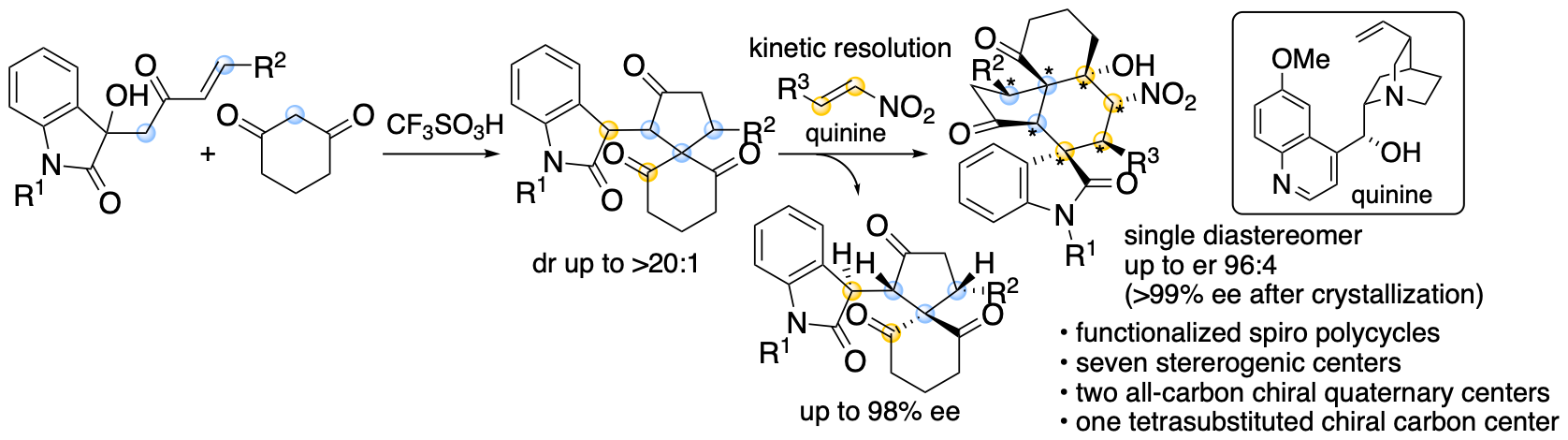
Spirooxindole derivatives are found in bioactive natural products. Functionalized molecules with spirooxindole cores including those with polycyclic systems should be useful in drug discovery efforts. We previously reported formal (4+1) cycloaddition and enantioselective Michael-Henry cascade reactions that provide spirooxindole polycycles via spiro[4,5]decane derivatives (Scheme 10) (Huang, Sohail, Taniguchi, Monde, and Tanaka, Angew. Chem. Int. Ed. 2017, 56, 5853). The reactions provided spirooxindole all-carbon polycyclic derivatives with seven stereogenic centers, including two all-carbon chiral quaternary centers and one tetrasubstituted chiral carbon center.
Scheme 10
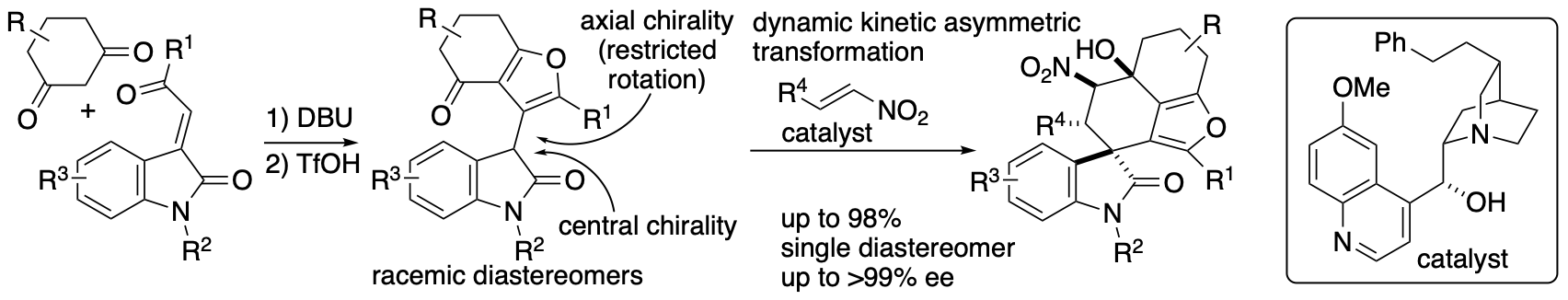
Whereas kinetic resolution reactions afford the products up to 50% yield, dynamic kinetic transformations can lead to the formation of the products up to 100%. We developed dynamic kinetic transformation-based diastereo- and enantioconvergent Michael-Henry reactions that afford spirooxindoles bearing furan-fused rings (Scheme 11) (Sohail and Tanaka, Angew. Chem. Int. Ed. 2021, 60, 21256). Dihydrobenzofuranone derivatives were synthesized in racemic diastereomer mixtures, and these were transformed to the spirooxindoles bearing furan-fused polycycles with high diastereo- and enantioselectivities through the dynamic kinetic process. Regardless the stereochemistry of the dihydrobenzofuranone derivative, all four isomers originated by central and axial chiralities were transformed to the single diastereomer product with high enantioselectivities.
Scheme 11
Usually, these complex, polycyclic molecules are synthesized by multi step routes. Our strategies allowed the access to the complex functionalized molecules including those as highly enantiomerically enriched forms in short routes. We are investigating the mechanisms of the reactions and the key factors leading to the formation of these products.
2.1.6. Control of chemical reactions using 1,3-cyclohexanedione derivatives as buffering molecules in non-aqueous solutions
Control of chemical reactions is necessary to obtain desired chemical transformation products and to prevent decompositions and isomerizations of molecules of interest. To control chemical reactions in aqueous solutions or to maintain conditions suitable for enzyme-catalyzed reactions and for storage of biological samples such as enzymes and antibodies, the use of buffers is common. We have introduced the "buffering" concept into the events that occur in non-aqueous solutions and have demonstrated that 1,3-cyclohexanedione derivatives have buffering functions in non-aqueous solutions (Scheme 12) (Sohail and Tanaka, Chem. Eur. J. 2020, 26, 222). We have demonstrated that 1,3-cyclohexanedione derivatives inhibit both acid-catalyzed and base-catalyzed isomerizations and decompositions in organic solvents. Using the buffering molecules that we have disclosed, there is no need to consider whether the decomposition and/or isomerization is caused by a base or an acid or which type of base or acid is causing the decomposition, isomerization, and/or racemization. Additionally, simply adding the buffering molecule to reaction mixtures can completely alter the pathways of the reactions.
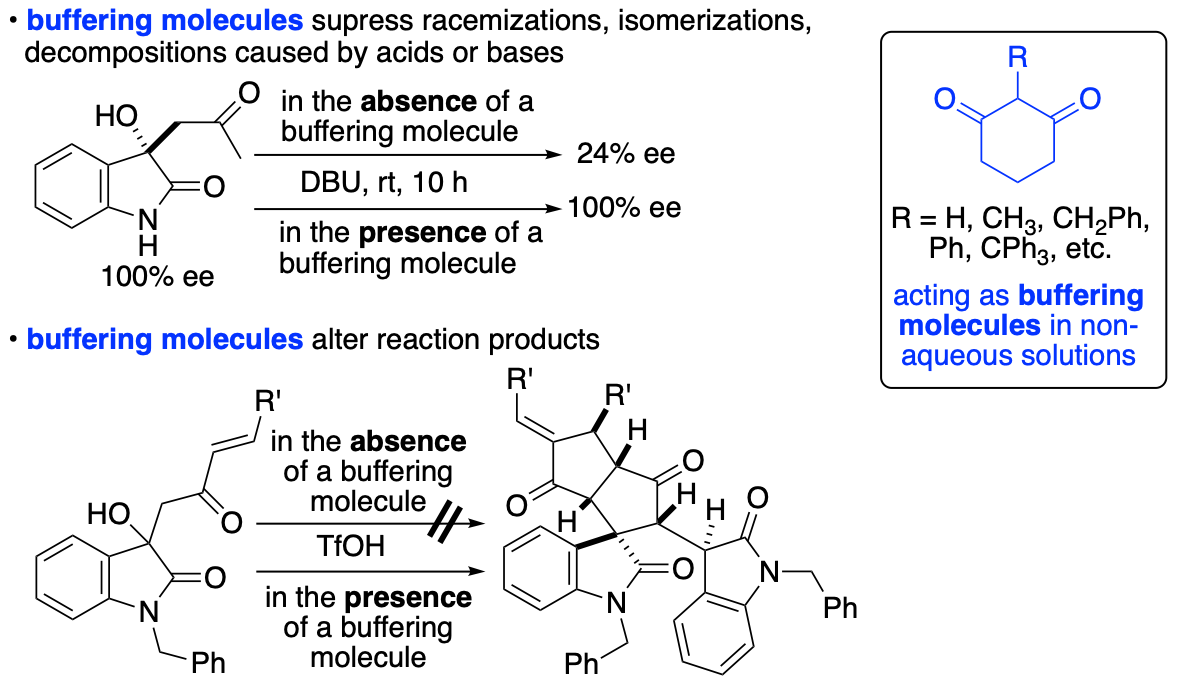
Scheme 12
As 1,3-cyclohexanedione derivatives have buffering functions in non-aqueous solutions, when 1,3-cyclohexanedione derivatives are used as reactants (such as shown in Scheme 10, for example), the loading amounts and the concentration of the 1,3-cyclohexanedione derivatives may affect the reaction rates and the outcomes. We have demonstrated that the limiting the loading amounts of reactant 1,3-cyclohexanedione in the acid-catalyzed formal (4+1) cycloaddition reaction enables the less use of the acid catalyst (Scheme 13) (Sohail and Tanaka, J. Org. Chem. 2023, 88, 10277). In certain cases, the reactions with higher concentrations of 1,3-cyclohexanedione were slower than those with lower concentrations. By minimizing the use of the cyclic 1,3-dione derivatives and by tuning the reaction concentration, the acid catalyst was reduced to 0.1 mol % to afford the desired products in high yields, and the reaction scope was expanded.
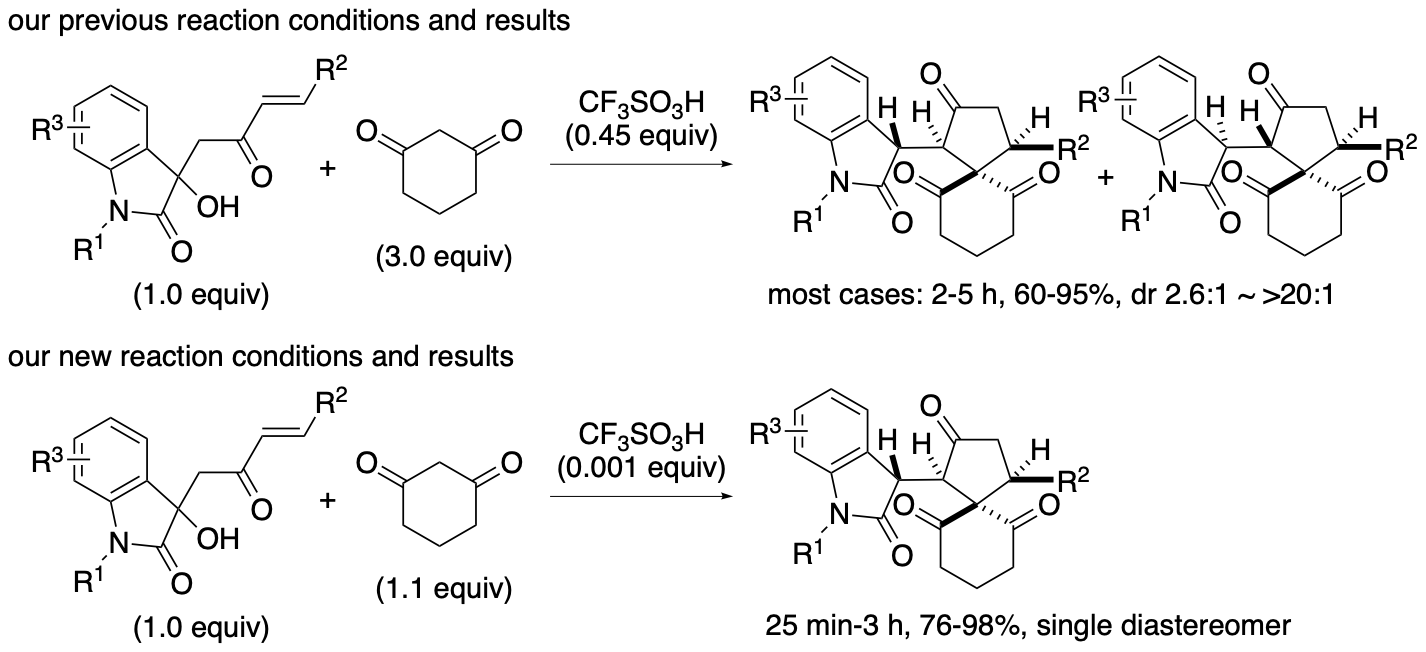
Scheme 13
High loading of 1,3-cyclohexanedione and/or a high concentration of 1,3-cyclohexanedione should shift the equilibrium of the binding of 1,3-cyclohexanedione with the acid or base toward the bound form (Scheme 14, AH = acid, B = base), inhibiting the interactions between the acid or base with the reactants and/or the intermediates that are necessary for the catalysis.
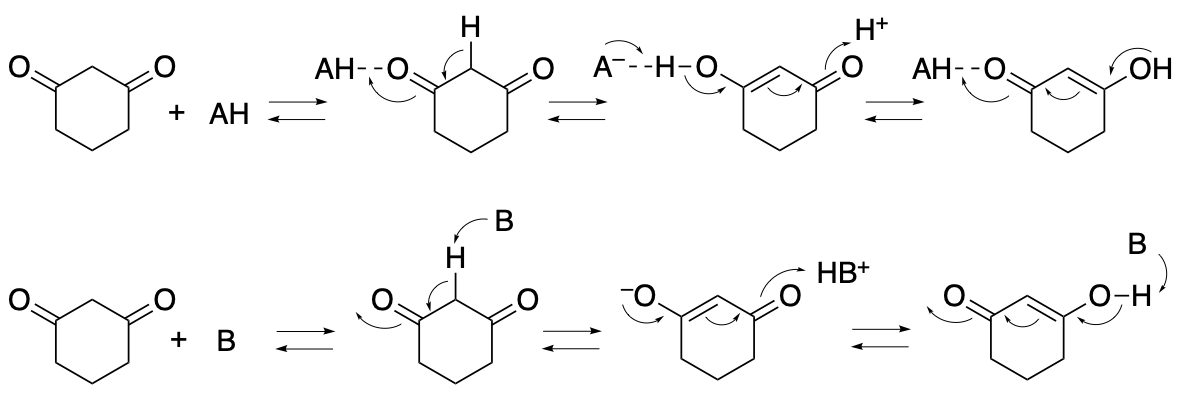
Scheme 14
We are expanding the concept for tuning the reactivities using enolizable cyclic 1,3-dione derivatives in organic solvents. We are also developing reaction systems and catalyst systems that use the buffering functions of cyclic 1,3-dione derivatives.
2.2. Protein and peptide modification reactions
Protein labeling methods are required for the synthesis of antibody-drug conjugates and other protein conjugates. Protein conjugates are important as therapeutics and are used in detection of molecules of interest. Conjugation reactions are also needed to create multifunctional protein- and peptide-based molecules. We have been developing protein and peptide modification reactions to synthesize conjugates. We have also been developing molecules that can be used for protein modification reactions at certain residues or targeted sites.
For example, we have developed chemical modification methods that use spirooxindole oxirane derivatives for the synthesis of protein- and peptide-conjugates (Scheme 15) (Chavan, Saze, and Tanaka, Adv. Synth. Catal. 2023, 365, 2171). In most cases, the epoxide group of the spirooxindole oxirane derivatives reacts to form a covalent bond with certain histidine residues and/or the N-terminal amino group of peptides and proteins. There are fewer histidine residues on protein surfaces than lysine residues and there is only a single N-terminal amino group per polypeptide chain. Thus, reactions at histidine residues and/or at the N-terminal amino group should result in more selective modification than reactions with lysine residues. The modification reactions of various proteins resulted in the formation of proteins modified at only one or two positions. Selected results of the modification reactions using the spirooxindole oxirane derivatives are shown in Figures 2 and 3.
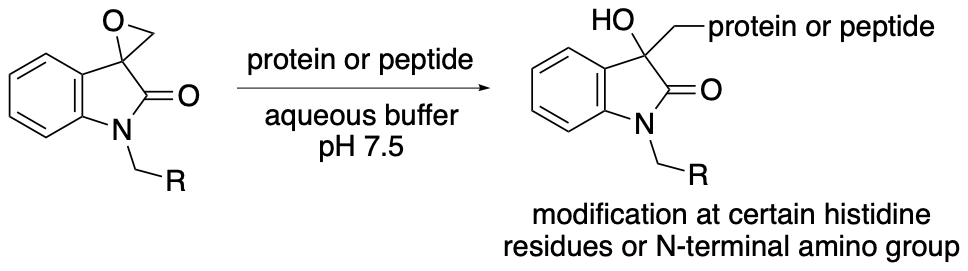
Scheme 15
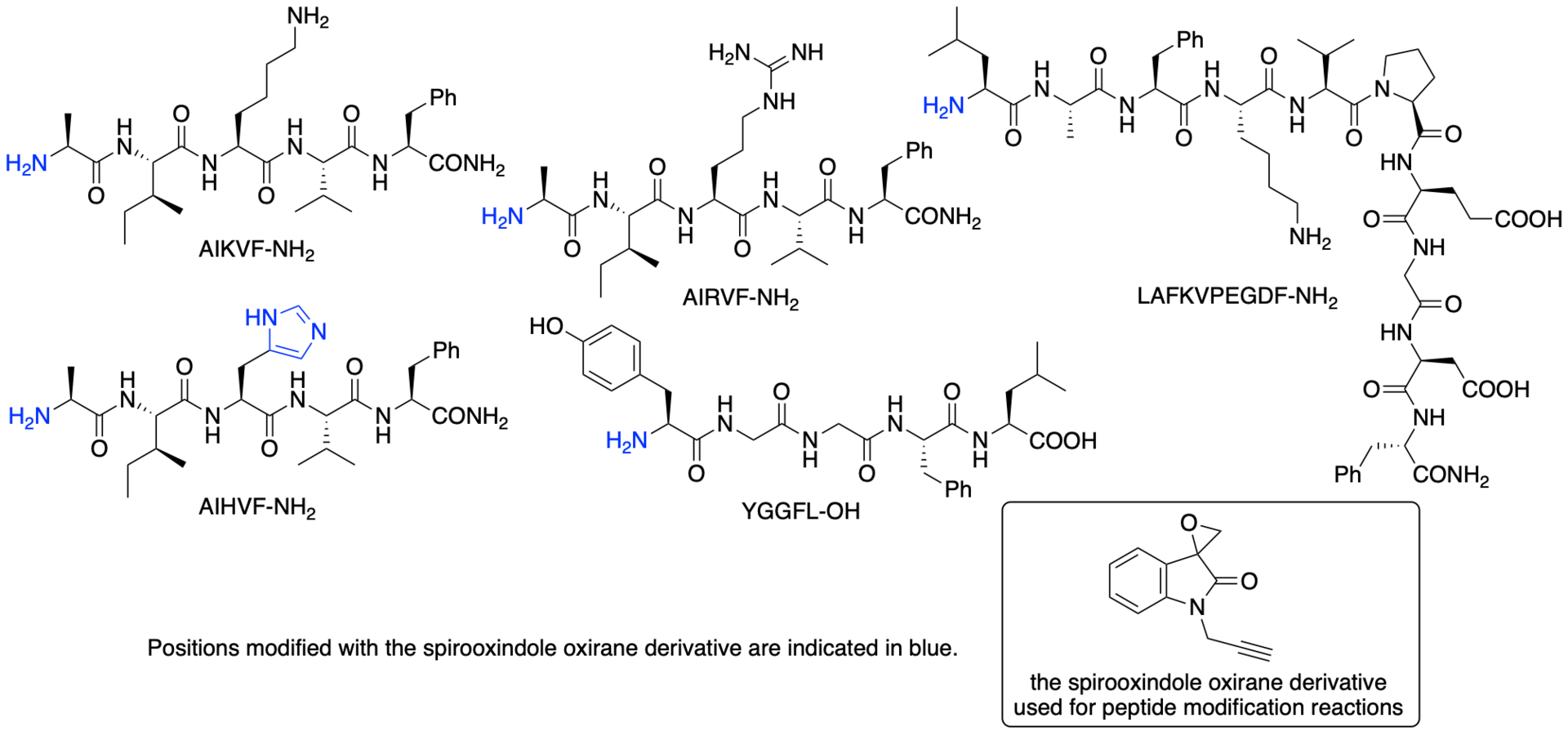
Figure 2
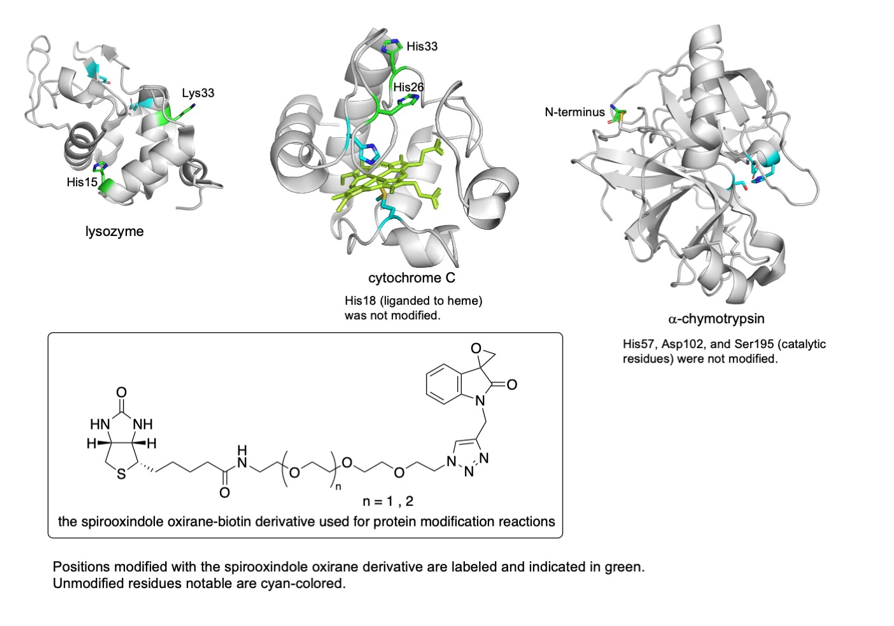
Figure 3
2.3. Search of biofunctional molecules
As described above, we have synthesized various functionalized molecules. In collaboration with researchers whose expertise is in biology and screening for biofunctional molecules, we have been searching new biofunctional molecules and drug leads.
3. Publications
3.1. Journals
- Bethi, V.; Bakhtawar, S.; Rupanawar, R. D.; Tanaka, F. Selective vinylogous aldol reactions of β-methyl-substituted cyclic enones with isatins catalyzed by DBU and Pyrrolidine to afford 3-substituted 3-hydroxyoxindoles. Asian J. Org. Chem. 2023, 12, e202300193, doi: 10.1002/ajoc.202300193.
URL: https://onlinelibrary.wiley.com/doi/full/10.1002/ajoc.202300193
- Sohail, M.; Tanaka, F. Limiting the loading of reactant 1,3-cyclohexanedione enables the use of less catalyst. J. Org. Chem. 2023, 88, 10277-10281, doi: 10.1021/acs.joc.3c00838.
URL: https://pubs.acs.org/doi/full/10.1021/acs.joc.3c00838
- Garg, Y.; Osborne, J.; Vasylevskyi, S.; Velmurugan, N.; Tanaka, F. 1,3-Diamine-derived catalysts: Design, synthesis, and the use in enantioselective Mannich reactions of ketones. J. Org. Chem. 2023, 88, 11096-11101, doi: 10.1021/acs.joc.3c01051.
URL: https://pubs.acs.org/doi/full/10.1021/acs.joc.3c01051
- Chavan, S. S.; Saze, H.; Tanaka, F. Chemical modification of peptides and proteins using spirooxindole oxirane derivatives. Adv. Synth. Catal. 2023, 365, 2171-2176, doi: 10.1002/adsc.202300578.
URL: https://onlinelibrary.wiley.com/doi/full/10.1002/adsc.202300578
- Chen, K.-L.; Tanaka, F. Organocatalytic enantioselective Mannich and retro-Mannich reactions and combinations of these reactions to afford tetrasubstituted α-amino acid derivatives. Org. Biomol. Chem. 2024, 22, 477-481, doi: 10.1039/d3ob01855e.
URL: https://pubs.rsc.org/en/content/articlelanding/2024/ob/d3ob01855e
3.2. Book Chapters
- Tanaka, F. The Bimolecular and Intramolecular Mannich and Related Reactions, In Additions to C-X π-Bonds (part 2), Yorimitsu, H. Ed. in Comprehensive Organic Synthesis, 3rd Ed, Knochel, P.; Molander, G. Ed.; Elsevier, in press, https://doi.org/10.1016/B978-0-323-96025-0.00016-8.
URL: https://doi.org/10.1016/B978-0-323-96025-0.00016-8
3.3. Oral and Poster Presentations
- Gupta, A.; Sohial, M.; Tanaka, F. Synthesis and applications of analogs of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), in Swiss Summer School on Organic Synthesis 2023, 2023.08.27-2023.08.31. (poster)
- Sohail, M.; Tanaka, F. Control of chemical reactions in non-aqueous solutions using buffering molecules, in The 15th International Kyoto Conference on New Aspects of Organic Chemistry (IKCOC-15), in Kyoto, Japan (2023), 2023.11.20-2023.11.23. (poster No. PB(D)-10)
- Osborne, J.; Garg, Y.; Tanaka, F. Enantioselective Mannich reactions of ketones catalyzed by new 1,3-diamine-derived catalysts, in The 15th International Kyoto Conference on New Aspects of Organic Chemistry (IKCOC-15), in Kyoto, Japan (2023), 2023.11.20-2023.11.23. (poster No. PB(D)-11)
- Mondal, S.; Tanaka, F. Direct Enantioselective Mannich Reactions of Pyruvates as Nucleophiles: Strategies for Controlling the Reactions using Organocatalysts, in The 15th International Kyoto Conference on New Aspects of Organic Chemistry (IKCOC-15), in Kyoto, Japan (2023), 2023.11.20-2023.11.23. (poster No. PB(D)-12)
- Osborne, J.; Garg, Y.; Tanaka, F. Enantioselective Mannich reactions of ketones catalyzed by new 1,3-diamine-derived catalysts, in the ACS Spring 2024, New Orleans, Louisiana & Hybrid, 2024.03.17-2024.03.21. (poster No. ORGN 3976783)
- Sohail, M.; Tanaka, F. Effects of reactant 1,3-cyclohexanedione on catalytic reactions: Limiting the loading enables the use of less catalyst in spiro ring formations, in The 144th Annual Meeting of the Pharmaceutical Society of Japan, Yokohama, Japan (2024), 2023.03.28-2023.03.31. (poster No. 29P-pm020)
- Chen, K.-L.; Tanaka, F. Catalytic enantioselective Mannich reactions of ketones with cyclic ketimino esters and their reverse retro-Mannich reactions to afford tetrasubstituted α-amino acid derivatives, in The 144th Annual Meeting of the Pharmaceutical Society of Japan, Yokohama, Japan (2024), 2023.03.28-2023.03.31. (poster No. 29P-pm024S)
4. Intellectual Property Rights
- Tanaka, F.; Yuvraj, Osborne, J. Novel compound or salt thereof, catalyst, method of preparing novel compound or salt thereof and method of preparing b-keto compound, patent filed, JP2023-085779, 2023.05.24.
- Tanaka, F.; Chavan, S., Use of spirooxindole oxirane derivative for modifying peptide and/or protein, patent filed, JP2023-113335, 2023.07.10.